Prader-Willi sendromu:
advertisement

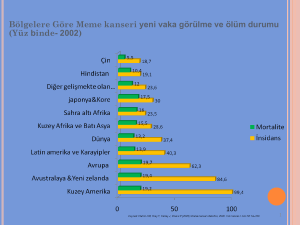
Free Copy for Web Publicatıon K. E. Gokay M.D., Ph.D. Prader-Willi sendromu: Prader-Willi sendromu başta zeka geriliği, duygulanım bozukluğu (duygusal dengesizlik), kaslarda güç kaybı ve hipotonus, kısa boylu cücelikle beraber iştah bozukluğuna bağlı morbid obezitenin gözlendiği nadir bir kalıtsal hastalıktır. Hastalık adını ilk kez 1956 yılında sendromu tarif eden üç hekimden almaktadır (Prader, Labhart ve Willi). Hastalığın görülme sıklığı değişkenlikler göstermekle beraber ortalama her 16,000 canlı doğumda bir (1:16000) olarak kabul edilebilir. Prader-Willi sendromunun moleküler genetik mekanizması babadan gelen (paternal) 15. (onbeşinci) otozomal kromozomun uzun kolunda 11 ila 13 segmentler arası bölgede mevcut olması gereken genetik materyalin yoksunluğudur (15q11-13). Vakaların % 70 ila 80’inde bu kayıbın bir mikrodelesyon sonucu oluştuğu bilinmektedir. Aynı kromozomun maternal (anneden gelen) kopyasında Prader-Willi sendromuna yol açan genler baskılanmış olduğundan (genomic imprinting) hastalık dominamt geçiş gösterir. İlginç olan, aynı mikro-delesyonun anneden gelen (maternal) 15. kromozomda olması ise tamamen başka bir hastalığa, Angelman sendromuna yol açmasıdır. Bu olgunun sebebi söz konusu kromozom bölgesinin maternal ve paternal genomda farklı şekillerde baskılanmış olmasındandır. Prader-Willi sendromu ve Angelman sendromu komşu bölgelerde yerleşik olsada farklı genlerin yoksunluğu sonucu oluşan dolayısıyla kendilerine has farklı klinik tablolar sergileyem iki ayrı sendromdur. Bir başka deyişle moleküler genetik tanı bakımından (genlerin komşuluğundan dolayı) benzerlik sergileselerde moleküler etiyoloji her iki hastalıkta biribirinden farklıdır. Angelman sendromunda rol oynayan temel gen bir ubiquitin ligazı kodlayan UBE3A geni iken Prader-Willi sendromunda rol oynayan primer genin mRNA işlenmesinde rol oynayan bir ribonükleoprotein olan (small ribonucleoprotein N) SNRPN geni olduğu rapor edilmiştir. Prader-Willi sendromunda moleküler genetik tanı, diğer tüm mikrodelesyon sendromlarında olduğu gibi, tercihen sitogenetik yöntemlerle yapılanlardır. Örneğin yüksek çözünürlükte prometafaz band boyaması ile yapılan karyotipleme ve/veya FISH (Fluorescent in-situ hybridization) tekniği ile 15q11-13 bölgesinin işaretlenmesi ile tanı konur. Angelman sendromunda olduğu gibi FISH tekniği ile hastaların en az dörtte üçünde (%75) lokus kaybı gösterilebilmektedir. Ancak ek olarak eksik parçanın paternal kopya olduğunun gösterilmesi de istenirse FISH analizini müteakip bu bölgenin STR (short tandem repeat) analizi yapılmalı ve STR profili anneden ve babadan elde edilen profillerle karşılaştırılmalıdır. Ancak kliniğin bariz olduğu çoğu olguda bu sadece akademik bir değer taşır. Prader-Willi sendromunda genetik analiz, hastalığın nadir görülmesinden ve henüz hastalarda tedaviye yönelik bir yöntem mevcut olmadığından dolayı, hastalara yalnızca tanı amaçlı testler olarak önerilmelidir. Hastalığın erken tanısında ve ayırıcı tanıda genetik analiz en kesin sonuç veren yöntemdir. Ancak prenatal ve yeni doğan tarama testleri veya taşıyıcılık testlerinin Prader-Willi sendromunda yeri henüz sınırlıdır.