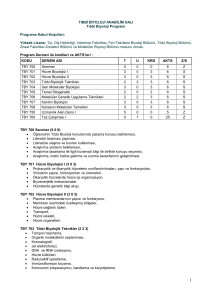
deneysel kafa travma modelinde montelukast sodyum`un oksidan
advertisement

T.C SAĞLIK BAKANLIĞI HAYDARPAŞA NUMUNE EĞİTİM VE ARAŞTIRMA HASTANESİ NÖROŞİRÜRJİ KLİNİĞİ Şef V. OP. DR. TAYFUN HAKAN DENEYSEL KAFA TRAVMA MODELİNDE MONTELUKAST SODYUM’UN OKSİDAN HASAR VE KAN BEYİN BARİYERİ ÜZERİNE OLAN ETKİSİ UZMANLIK TEZİ Dr. NECAT BİBER İSTANBUL-2008 I. TEŞEKKÜR Her türlü fedakarlığa katlanarak bugünlere gelmemi sağlayan aileme başta olmak üzere; nöroşirürji eğitimim süresince bilgi, birikim ve desteğini esirgemeyen değerli hocam Prof. Dr. M. Zafer Berkman’a, bilgi ve tecrübelerinden faydalandığım şef yardımcım, ve Op. Dr. Tayfun Hakan’a; mesleki gelişmemde katkıları olan uzman doktorlar; Doç. Dr. Kaya Kılıç, Op. Dr. Metin Orakdöğen, Op. Dr. Hakan Somay, Op. Dr. Mehmet Erşahin, Op. Dr. Hasan Çelik, Op. Dr. Aydın Aydoseli, Op. Dr. Cezmi Ç. Türk, Op. Dr. Erdoğan Ayan’a; ve bir dönem birlikte çalıştığımız kliniğimiz uzman doktorları; Op. Dr. Cumhur Özdoğan, Op. Dr. Serdar Armağan, Op. Dr. Selhan Karadereler ve Op. Dr. M. A. Göğüsgeren’e, tezimin bu hale gelmesinde önemli katkılarından dolayı Marmara Üniversitesi Eczacılık Fakültesi Farmakoloji AD.’da görev yapan Yrd. Doç. Dr. Hale Zerrin Toklu başta olmak üzere Prof Dr. Gül Dülger, Prof. Dr. Göksel Şener ve tüm çalışanlarına; elektron mikroskopisi incelemelerinde katkıda bulunan İstanbul Üniversitesi İstanbul Tıp Fakültesi Histoloji ve Embriyoloji AD. Prof Dr. Seyhun Solakoğlu, Asistan Dr. Mine Gültomruk’a; eğitimim süresince görev yapan hastanemiz başhekimleri, doktor ve personellerine; birlikte çalışmaktan büyük mutluluk duyduğum ve herşeyimizi paylaştığımız sevgili asistan arkadaşlarıma, servisimiz ve ameliyathanemiz hemşire ve personeline teşekkür ederim. Her zaman sevgi ve desteklerini hissettiğim, sevili eşim Selgin, oğlum Kemal ve kızım Defne’ye sonsuz teşekkürler…. Dr. Necat BİBER I II. İÇİNDEKİLER I) TEŞEKKÜR II) İÇİNDEKİLER III) KISALTMALAR 1. ÖZET....................................................................................................................1 2. GİRİŞ VE AMAÇ................................................................................................2 3. GENEL BİLGİLER............................................................................................4 3.1. Kafa travması................................................................................................4 3.1.1. Tarihçe............................................................................................4 3.1.2. Deneysel kafa travması modelleri.................................................5 3.1.3. Epidemiyoloji..................................................................................8 3.1.4. Sınıflama.........................................................................................9 3.1.5. Travmatik Beyin Yaralanmalarında Fiziksel Mekanizmalar.11 3.1.6. Primer Beyin Yaralanmaları Tipleri..........................................12 3.1.6.1. Skalp Laserasyonları....................................................13 3.1.6.2. Kafa Kırıkları................................................................13 3.1.6.3. Travmatik intrakranyal kanamalar............................13 3.1.6.4. Kontüzyon ve Laserasyon............................................15 3.1.6.5. Diffüz Aksonal Yaralanma..........................................16 3.1.7. Sekonder Beyin Yaralanmaları..........................................................................17 3.1.7.1. Sebepleri..........................................................................17 3.1.7.2. Fizyopatotoloji...............................................................18 3.2. Beyin Ödemi................................................................................................22 3.2.1. Ödemin sınıflandırılması.............................................................22 3.3. Kan Beyin Bariyeri.....................................................................................24 3.4. Oksidan Hasar ve Savunma Mekanizmaları............................................25 3.4.1. Serbest radikaller ve Oksidan hasar..........................................26 3.4.1.1. Serbest Radikallerin Fizyopatolojik Olaylardaki Rolü............................................................28 3.4.2. Endojen Antioksidan Sistem.......................................................29 II 3.5. Sistenil Lökotrienler...................................................................................30 3.6. Montelukast Sodyum..................................................................................32 3.6.1. Farmakokinetik Özellikleri.........................................................32 3.6.2. Farmakolojik etkileri...................................................................33 4. GEREÇ VE YÖNTEM....................................................................................35 4.1. Deney Hayvanları.......................................................................................35 4.2. Kafa Travması Modeli...............................................................................35 4.3. Nörolojik Muayene.....................................................................................37 4.4. Ölçülen Parametreler.................................................................................39 4.4.1. Beyin Ödemi...............................................................................39 4.4.2. KBB Geçirgenliği.......................................................................39 4.4.3. Oksidan Hasar Ve Savunma.....................................................42 4.4.4. MDA............................................................................................42 4.4.5. GSH.............................................................................................43 4.4.6. MPO............................................................................................44 4.4.7. Na+ K+-ATPaz Tayini................................................................46 4.4.8. Histolojik incelemeler ve perfüzyon fiksasyonu....................48 4.5. İstatistiksel Değerlendirme........................................................................49 5. BULGULAR.......................................................................................................50 5.1. Nörolojik Muayene.....................................................................................50 5.2. MDA ve GSH..............................................................................................51 5.3. MPO ve Na-K-ATPaz.................................................................................52 5.4. Kan beyin bariyeri geçirgenliği.................................................................53 5.5. Histolojik incelemeler.................................................................................55 6. TARTIŞMA VE SONUÇ..................................................................................56 7. KAYNAKLAR...................................................................................................61 8. ETİK KURUL RAPORU..................................................................................70 III III. KISALTMALAR AD Atopik Dermatit ANOVA Analysis of varience, varyans analizi testi AQP4 Akuaporin–4 AR Alerjik Rinit ATP Adenozin trifosfat BOS Beyin Omurilik Sıvısı BBT Bilgisayarlı Beyin Tomografi CysLT Sisteinil lökotrien DNA Deoksiribonükleik asit DTNB Ditiyo nitro benzen EB Evans mavisi FLAP 5-LO aktive eden protein G6PD Glukoz–6 fosfat dehidrojenez GKS Glasgow Koma Skalası GPCR G-proteini ile eşleşmiş spesifik reseptörler g Gram GSH Glutatyon GSH-Px Glutatyon peroksidaz GSSG İndirgenmiş glutatyon HETAB Hekzadesiltrimetil-amonyum bromid İL İnterlökin ip İntraperitoneal iv İntravenöz KBB Kan beyin bariyeri KİB Kafa İçi Basınç LT Lökotrien MDA Malondialdehid MPO Myeloperoksidaz MRG Magnetik Rezonans Görüntüleme MS Multipl Skleroz IV NADPH Nikotinamid adenin dinükleotid NMDA N-metil-D-aspartat NO Nitrit Oksit NO2+ Nitronyum iyonu ONOO- Peroksinitrit PARP–1 Poli(ADP-riboz) polimeraz PARS Poli(ADP-riboz) sentaz ROM Reaktif oksijen metabolitleri SKA Serebral kan akımı SOD Süperoksit dismutaz SPB Serebral perfüzyon basınç SSS Santral sinir sistemi TBA Tiyobarbitürik asit TBY Travmatik Beyin Yaralanması TCA Trikloroasetik asit TNF-α Tümör nekroz faktör-alfa VEGF Vasküler endotelyal büyüme faktörü V 1. Özet Nöropatolojik hastalıkların temelinde serbest radikallerin aşırı miktarda üretiminin rol oynadığı gösterilmiştir. Oluşan serbest radikaller hücre yapısındaki protein, lipid ve DNA gibi hücresel elemalarda hasara neden olmaktadır. Montelukast, sisteinil lökotrien (CysLT1) reseptörlerini bloke ederek enflamasyonu baskılamaktadır. Montelukastın antienflamatuvar etkilerinin yanısıra antioksidan etkileri olduğu da ileri sürülmektedir. Bu çalışmada 1m yükseklikten 300 g ağırlık düşürülerek oluşturulan sıçanlardaki kafa travmasında montelukastın kan beyin bariyeri geçirgenliği, beyin ödemi, malondialdehid (MDA), glutatyon (GSH), miyeloperoksidaz (MPO), Na/K-ATPaz pompası, histomorfolojik ve nörolojik değişikliklere etkileri araştırılmıştır. Travma oluşturulmasından 48 saat sonra yapılan nörolojik muayenede travma grubunda muayene skorlarında kötüleşme saptanmıştır. Aynı zamanda ödemin göstergesi olarak beyin su miktarı artarken, kan beyin engeli geçirgenliği de artmıştır. Montelukast tedavisi ödeme etkisiz olmakla birlikte, kan beyin engelini korumuş, MDA ve MPO düzeylerini de anlamlı olarak düşürmüştür. Sonuç olarak, bu çalışmada montelukastın kapalı kafa travması modelinde görülen beyin hasarına karşı koruyucu etki gösterdiği bulunmuştur. Anahtar Kelimeler: Beyin, Beyin ödemi, Kan beyin engeli, Kapalı kafa travması, Montelukast, Na/K-ATPaz Oksidatif stress; 1 2. Giriş ve Amaç Kafa travması 1–44 yaşları arasında ölümlerin en sık sebeplerinden ve endüstrileşmiş toplumlarda 45 yaşından küçük hastalarda nörolojik hasara bağlı sekellerin başta gelen nedenidir. Asıl ölüm sebebi kafa travması ile birlikte olan primer yaralanmadır ve travma sonrası birkaç dakika veya saatler içinde gelişen sekonder yaralanma ile mortalite ve morbidite artmaktadır. Kafa travmasının akut etkilerini ya da geç sekonder hasarını azaltmak için yeni ufuklar açabilecek ve yol gösterici olacak tedavi stratejileri araştırımak için mekanik beyin yaralanmasını temsil eden hayvan modelleri geliştirilmiştir. Şiddetli kafa travmasında fokal kitle lezyonu olmaksızın travmaya sekonder yaygın beyin hasarı olabilir. Primer yaralanma, fokal veya difüz yaralanmaya neden olan çarpışma esnasında kafatasına uygulanan mekanik güçlerin sonucu iken, sekonder beyin zedelenmesi esas olarak zarar verici hareket tarafından başlatılan, resusitasyonun sırasında erken hipoksi ve hipotansiyonun ana risk faktörlerini oluşturduğu bir dizi karmaşık süreçleri temsil etmektedir. Sekonder olaylar doğal immün yanıtı, kompleman aktivasyonu ile intrakranyal kompartman içinde büyük bir konakçı enflamatuvar yanıtını aktive ederek nöroenflamasyonu başlatır. Nöroenflamasyon, kandaki lökositlerin kan beyin engelini aşarak subaraknoid boşluğa doğru geçmesine bağlıdır. Nötrofil ve diğer bağışıklık hücrelerinin aktive olması sonucu açığa çıkan proenflamatuvar sitokinler, kemokinler ve kompleman anafilatoksinler nöroenflamasyonda rol oynayan endojen mediyatörler arasında sayılabilir. Ortaya çıkan bu ikincil immün yanıt ile travmaya sekonder olarak serebral iskemi ve beyin ödemi de gelişebilir. Arişidonik asidin 5-lipooksijenaz metoboliti olan sisteinil lökotrienlerin (CysLT) iskemi, travma, tümör, multiple skleroz (MS), ensefalomyelit ve yaşlanma gibi çeşitli santral sinir sistemi (SSS) hastalıklarında üretimi artar. CysLT’lerin beyinde birikimi serebral iskemide anahtar rol oynayabilir. Nötrofiller iskemik alana göç ettiğinde reaktif oksijen ürünleri, proteazlar, elastazlar, myeloperoksidaz (MPO), sitokinler ve doku zedelenmesinde yer alan çeşitli diğer sitokinleri açığa çıkarırlar. Montelukast, eozinolotik enflamasyonu bu hücrelerden enflamatuvar sitokin salınımını inhibe ettiği gösterilmiştir. 2 Bu çalışmanın amacı kafa travmasının patojenezinde rol oynayan sekonder mekanizmalardan beyin ödemi, kan beyin bariyeri geçirgenliği ve oksidan hasar araştırılması ve başka modellerde (iskemi-reperfüzyon, astım gibi) antioksidan ve antienflamatuvar etkileri gösterilmiş olan montelukastın bu travma modelinde olası koruyucu etkilerinin incelenmesidir. 3 3. Genel Bilgiler 3.1. Kafa travması 3.1.1. Tarihçe Nöroşirürjinin kafa travmasının tedavi edilmesi ile başladığı düşünülmektedir. Okların sebep olduğu birleşik çökme kırıkları eski Çin hanedanlıklarından beri bilinmektedir. Travmatik pıhtıların delik açılarak boşaltıldığında iyileştiği, böylelikle hastaların yaşadığı İnkalar zamanında bulunmuştur. Hipokrat, kafa travmalarını sınıflayarak bazılarında delik açılmasını önermiştir (53). İkinci Dünya Savaşı’ndan sonra kafa yaralanmalarının tedavisinde iki büyük gelişme olmuştur. Bunlar, yoğun bakım yöntemleri ile anestezist, kulak burun boğaz uzmanı ve nöroşirürjiyen gibi uzmanların hastayı birlikte multidisipliner bir yaklaşımla ele almasıdır. Böylece, şiddetli yaralanmalarda bile hastaların daha hızlı ve kolay iyileşmesi sağlanmıştır (53). Bir başka dramatik ilerleme de bilgisayarlı beyin tomografisi (BBT) tetkikidir. 1970’lerin ortalarında BBT’in uygulamaya girmesi, yaşamı tehdit eden kanama olduğundan kuşkulanılan hastalara araştırma amaçlı açılan burr holeler açılmasına son vermiştir. BBT’nin ABD’de çok yaygın olması hem ılımlı hem de şiddetli kafa travmasının araştırılması ve tedavi edilmesinde erindeki eğilimleri değiştirmiştir. Bu durum sadece nöroşirürji ve yoğun bakım ünitelerinde görülenden farklı olarak kafa travmalı hastalarla hastane çapında bir organizasyonla ilgilenilmesine yol açmıştır. Manyetik rezonans görüntüleme (MRG), BBT’de görülmeyeni görünür hale getirerek lezyonlar hakkındaki bilgimizi genişletmektedir. MRG özellikle klinik standartlara göre sadece hafif yaralı hastalarda beyin hasarının ne kadar yaygın olduğunu gösterebilmektedir. Denny-Brown ve Russell’in Oxford’da 2. Dünya Savaşı’nın ilk yıllarında yaptıkları çalışmada travma esnasında maymunun kafası serbest olduğunda beyin hasarının daha geniş olduğunu göstermişlerdir. Daha sonra Generalli ve ark.ları da deneysel primat çalışmalarında bu çalışmaya genişleterek doğrulamışlardır. Oxford’daki fizikçi Holbourn 1940’ larda beynin jelatin modelleriyle deneyler yapmış ve ak 4 maddede bugün diffüz aksonal yaralanma denilen yırtılma lezyonlarını taklit etmiştir (28, 53). Philadelphia ve Glasgow’daki takımların birlikte yaptıkları araştırmaların sonucu olarak insanlarda bulunan tüm lezyonlar değişen dereceler, süreler ve akselerasyon ve deselerasyon süreleri ile deneysel olarak yeniden oluşturulabilir. Sinir lifleri ve kan damarlarına etki eden, yırtılmaya neden olan kuvvetlerin çarpma veya primer hasara yol açan temel mekanizmalar oldukları konusunda artık kuşku yoktur. Bu deneyler aksonal hasarın sürekli olabileceğini göstermiştir. Bunun kalıcı bir lezyona doğru ilerleyip ilerlemeyeceği farmakolojik girişimlerle etkilenebilen faktörlere dayanabilir. Bu, başlangıçta hasarın maksimal ve geri dönüşümsüz olduğu varsayımına dayalı çarpışmaya bağlı beyin hasarı felsefesini değiştirmiştir. Ölen hastaların birçoğunda hipoksik veya iskemik beyin hasarı ve kafa içi basınç artışının olması, yoğun solunum bakımı ve diğer tıbbi önlemlerden sağlanan faydayı açıklamaktadır. Yıllarca KİB’ı, steroid ilaçlar, ozmotik ajanlar, hiperventilasyon ve hipotermi ile beynin metabolik hızını düşürerek kontrol altına alınmaya çalışılmıştır. Hasta nöroşirürjikal bakım aldıktan sonra arteryel hipotansiyon ve KİB artışı epizodlarıyla sık karşılaşıldığı kesinleşmiştir. Bu epizodlar, yaralanma sonrasındaki erken evrelerde ortaya çıkan epizodlar ile birlikte, sekonder iskemik hasarın neden sık olduğunu açıklamaktadır. Artık böylesi kötü sonuçları olabildiğince çabuk tespit edip düzeltmek için monitör sistemleri kullanılmaktadır. Monitörizasyonların ve terapötik girişimlerin çoğunun riskleri olduğu bilinmektedir. Sonucun daha iyi olmasının modern yoğun bakım bileşenlerinden (sıklıkla “agresif tedavi” denilen) bazılarının doğruluğunu gösterip gösteremeyeceği kesin değildir. 3.1.2. Deneysel kafa travması modelleri Kafa travmasında hayvan modelleri, insandaki kafa travmasının primer ve sekonder sebeplerini araştırmak ve insan beyni mevcut sinir hasarını onarmaya yönelik çeşitli tedavi yöntemlerini geliştirmek için yapılmaktadır (19). Deneysel model seçimi araştırmanın amacına göre olmalıdır. Modellerin temel amacı nekrotik ve apitotik nöronal hücre ölümünü ortaya koymak ve travmaya sekonder gelişen davranış değişikliği, fizyolojik, metabolik, kan doku değişiklikleri, kan beyin 5 bariyeri geçirgenliği, enflamatuvar ve immunolojik cevaplar hakkında bilgi sahibi olmaktır (19). Hücre ölümünün temel sebebleri iyonik dengelerin bozulması, ortaya çıkan serbest radikaller, enflamatuar ve immunolojik cevap, ekstitatör aminoasitlerin salınması, nörotransmitter ve nöromodülatör sistemlerin bozulması olarak sayılabilir (19). Deneysel kafa travmaları, Denny-Brown Russell’in çalışmalarıyla akselerasyon konküzyon ve perküsyon-konküzyon olmak üzere iki kategoriye ayırmıştır (28). Mekanik kuvvet amplitüdüne, süresine, hızına ve akselerasyonuna bağlı olarak hem dinamik hem de statik beyin travmasına neden olur. Statik modellerdeki mekanik kuvvet belirlenmiş amplitüd ve süreye sahipken, hız ve akselerasyon önemli değildir. Statik modeller doğal olarak genellikle yaralanmada yer alan morfolojik ve fonksiyonel süreçlere odaklanır. Diğer taraftan ise amplitüd, süre, hız ve akselerasyonu iyi belirlenmiş mekanik kuvvet, dinamik beyin yaralanmasına neden olur. Dinamik beyin yaralanması direkt ve indirekt beyin yaralanması olarak alt gruplara ayrılır. İndirekt dinamik beyin yaralanmasında mekanik kuvvet genellikle tüm vücuda yöneliktir, salınım basınç dalgalarının kinetik enerjisi vücudu geçerek etkilerini beyin dokusu üzerinde açığa çıkarırlar. Travmatik beyin yaralanmasının in vivo deneysel hayvan modellerinin şematik temsili aşağıdaki Şekil 1 de gösterilmiştir. 6 STATİK MEKANİK KUVVET İNDİREKT DİNAMİK DİREKT ÇARPMANIN OLMADIĞI BAŞ AKSELERASYONU ÇARPIŞMA PENETRAN YARALANMA/ DİREKT BEYİN DEFORMASYONU (Baş Hareketi) ZORLANMIŞ • Lateral Sıvı Perküsyonu • Kontrollü Kortikal Çarpışma • Fokal Kortikal Kompresyon • Yüksek Hızlı Titreşimli Hasar ZORLANMAMIŞ • Yüksek Hızlı Titreşimli Hasar PENETRAN OLMAYAN YARALANMA/ ÇARPIŞMA AKSELERASYONU (Baş Hareketi) ZORLANMIŞ • Kontrollü Kontüzyon • Ağırlık Düşürme Modelleri ZORLANMAMIŞ • İlkel Çarpışma Akselerasyon Modelleri • Koyun türü Çarpışma Modeli (Baş Hareketi) ZORLANMIŞ • Minyatür domuzlar • Tavşanlar • Sıçanlar ZORLANMAMIŞ Primatlar Şekil:1 Deneysel hayvan modellerinin şematik temsili (19) 7 Marmarou’nun impakt akselerasyon modeli, diğer modellere göre uygulanması kolay, ucuz, kontrol edilebilir olması nedeniyle popülerdir. Hem apopitik hem de nekrotik hücre ölüm hasarı ortaya çıkarır. Histopatolojik yaygın bilateral nöron, akson, dentrit ve mikrovasküler hasarı oluşturur. Özellikle korpuz kallozum, internal kapsülde, optik traktusta, serebral ve serebellar pedinkülde ve beyin sapında diffüz aksonal yaralanma olur (67). İlk 4 saatte serebral kan akımında azalma ve intrakranyal basınçta artma ile sekonder otoregülasyon bozukluğu oluşur. İlk saatlerde vazojenik ödem daha sonra yaygın hücresel şişme meydana gelir. İlk 20 dakikada başlayan ve 24 saate kadar devam eden beyin ödemi oluşur. İlk 4 saatden sonra ise ödemle ilişkili olarak kan beyin bariyeri geçirgenliğinin bozulduğu gösterilmiştir (19). 3.1.3. Epidemiyoloji Kafa travmaları toplumda oldukça sık görülen önemli saglık ve sosyo-ekonomik problemlere neden olmaktadır. Travmatik beyin yaralanması (TBY) ABD’de tahminen direkt ve indirekt maliyeti 56 milyar doları aşan önemli bir sakatlık nedenidir. Ayrıca adli, tıbbi ve cerrahi açılardan da önemli bir konudur (45). Kafa travmalarının epidemiyolojisi sosyo-ekonomik seviye farklılıklarına, yaş, ırk ve cinsiyete göre değişim göstermektedir. Yapılan araştırmalarda kafa travmalarında 15-25 yaş grubunun risk yüzdesinin fazla oldugu görülürken, kafa travması insidansı 25-60 yaş grubunda düsme egilimine girmekte, 60 yaşından sonra ise tekrar yükselmektedir. Kadın/erkek oranı ise 1/2-1/2.8 arasında degişmektedir. Sosyoekonomik seviyesi düşük toplumlarda kafa travması görülme oranı daha yüksektir (60). Genel travmaya bağlı ölümlerin % 50’si kafa travmasına bağlıdır. Kafa travmasına bağlı ölümlerin % 50’si hastaneye ulaşmadan gelişmekte, hastanedeki ölümlerin 2/3’ü ise ilk 24 saat içinde olmaktadır. Bu ölümlerin 1/3’ü primer beyin hasarına bağlı iken, sekonder beyin hasarına bağlı ölümlerin % 90’ı kontrol edilemeyen KİB artısı ile ilişkilidir (52). Bebekler, çocuklar ve genç erişkinler TBY açısından yüksek risk altında olsalar da 75 yaşın üstündekilerin yaralanma yüzünden hastaneye yatmaları ve ölmeleri daha olasıdır (45). 8 Son yıllarda, kafa travmalarının fizyopatolojisi hakkındaki bilgilerde artmasına, yoğun bakımdaki hasta izleme ve bakım tekniklerindeki gelişmelere bağlı olarak mortalite oranı % 20 düzeylerine kadar azalmış ve prognozda belirgin düzelmeler gözlenmiştir (50). 3.1.4. Sınıflama Kafa travmasının evrensel olarak bilinen ve kabul edilen ortak bir sınıflaması yoktur. İdeal sınıflama hangi hastaya BBT tetkiki yapılması, hangi hastanın gözleme alınması, hangi hastaya acil cerrahi girişimde bulunulması gerektiğini ve hangi hastada tehlikeli KİB artışı gelişeceğini güvenilir şekilde ön görebilecek esnekliğe sahip olmalıdır (96). Sınıflamada olması gereken bilgiler intrakranyal anormalliklerin tipi, lokalizasyonu ve büyüklüğü ile aynı zamanda kaza öyküsü, kafaya çarpmanın şiddeti, nörolojik disfonksiyonun derecesi, zamansal seyri, hastaneye gelmeden önceki hali ile ilişki olmalıdır (96). Varolan sınıflama sistemleri kategorik, matematiksel, özel amaca yönelik gruplamalar, değişkenlerin birleşimi olarak ayrılabilir. Matematiksel sınıflamalar arasında en sık evrensel olarak kabul edilen şiddetin derecesini gösteren Glasgow Koma Skalası (GKS) kullanılmaktadır (96). 9 GLASGOW KOMA SKALASI (GKS) Spontan Açık 4 Sözlü emirler ile açar 3 Ağrı ile açar 2 Yanıt yok 1 Sözlü emirlere uyuyor 6 Ağrılı emrleri lokalize eder 5 MOTOR Fleksör yanıt 4 YANIT Dekortike yanıt 3 Desebre yanıt 2 Yanıt yok 1 Oryante 5 VERBAL Konfüze konuşma 4 YANIT Anlamsız Kelime 3 Anlamsız ses 2 Yanıt yok 1 GÖZ AÇMA Tablo : 1 Glasgow koma skalası (99). Liege grubu tarafından GKS’a beyin sapı refleksleri eklenmiştir. GKS-Liege Sınıflaması Beyin Sapı refleksleri Fronto-orbiküler 5 Vertiklaokülovestibüler 4 Pupiller ışık refleksi 3 Horizontal okülovestibüler 2 Okulokardiyak 1 Tablo: 2 GKS-Liege Sınıflaması (14) 10 Bu sınıflamaya göre (44): • Hafif-Minör kafa travması; (% 75–80) GKS 14–15, geçici bilinç kaybı (5 dakikadan az), fokal nörolojik defisit yok. • Orta kafa travması; (% 10-20) GKS 9-13, fokal nörolojik defisit +/-, bilinç kaybı (5 dak. fazla ) primer ve sekonder yaralanma eşlik eder yoğun bakım gerekebilir, sekel kalabilir. • Ağır-şiddetli kafa travması; (% 10–20) GKS 5- 8, komatöz, yoğun bakım gerekir, mortalite ve morbidite yüksektir, yaşayanlarda sekel vardır. Travmatik beyin yaralanmaları karmaşık bir süreç olup primer yaralanma, primer yaralanmanın gelişmesi, sekonder ve ek yaralanma ve rejenerasyonu içine alan üst üste binen fazlardan oluşur (82) . Primer beyin yaralanmaları, travma anında ya travmanın direkt etkisi sonucu beyin parankiminde ya da akselerasyon ve deserelasyon kuvvetlerine bağlı uzun beyaz cevher traktuslarında meydana gelir. Beyin parankimasına olan direkt travma, beynin kemik protuberanslara çarpmasının veya beynin kemik fragmanlar ya da yabancı cisim tarafından penetrasyonu sonucu oluşur. Akselerasyon-deselerasyon kuvvetleri uzun beyaz cevher traktuslarında kopup ayrılmaya neden olarak aksonal bozulma ve sekonder hücre ölümlerine yol açarlar (32). Sekonder beyin yaralanmaları başlangıç travmasını takip eden sistemik ve intrakranyal olaylardır, primer travmaya bir yanıt olarak meydana çıkarlar ve nöronal harabiyet ile hücre ölümüne yol açarlar. Primer travma tarafından tetiklenen enflamatuvar olaylar bir dizi biomoleküler değişikliklerin bir sonucu olup mikrosirkülasyonun bozulmasına ve nöronal bütünlüğün ortadan kalkmasına yol açar. Dolayısıyla sistemik hipotansiyon, hipoksi, hiperkapni, artan intrakranyal basınç gibi sekonder yaralanmalar orijinal beyin travması ya da multipl travmalar tarafından başlatılan bir seri fizyolojik değişikliklerin biyokimyasal sonuçlarıdır (32). 3.1.5. Travmatik Beyin Yaralanmalarında Fiziksel Mekanizmalar Travmatik beyin yaralanmalarında fiziksel mekanizmalar aşağıdaki gibi sınıflandırılırlar: 11 • Darbenin yüklenmesi (örn. başın solid bir cisimle belirli bir hızla çarpışması). • Tepkisel yüklenme (örn. belirgin fiziksel bir temas olmaksızın ani hareket). • Statik ya da hemen hemen statik yüklenme (örn. yüklenmede oluşumun hızının etkisinin belirgin olmadığı durumlar). Darbe yüklenmeleri, temas kuvvetleri ile atalet kuvvetlerinin bir bileşimidir. Atalet kuvvetleri, bir temas kuvveti olsun olmasın başın harekete geçmesi sonucu başın ivme kazandırması sonrası ortaya çıkan kuvvetlerdir. Temas kuvvetleri baş istirahat halinde, hareketsiz iken başa yüklenen darbe sonucu ortaya çıkar. Statik veya hemen hemen statik yüklenme oldukça ender olup yavaş hareket eden bir objenin rijit bir yapıda olan başı sabit bir durumda yakalaması ve tedrici sıkıştırması sonucu meydana gelir. Kafatası deformasyonuna hatta ölüme yol açabilen pek çok kommünike kırık ile karakterizedir. Temas veya atalet kuvvetleri beyin dokusunu yapısal toleransının ötesinde gererek, zorlayarak yaralanmaya yol açar. Gererek zorlama uygulayan mekanik bir kuvvetin deformasyona uğrattığı 3 temel tip doku deformasyonu mevcuttur: A) Sıkıştırıcı tarzda (doku kompresyonu) B) Gerilme tarzında (dokunun gerilmesi-esnemesi) C) Yırtılma tarzında (doku distorsiyonu) olup bir dokunun diğeri üzerinde kayması sonucu. (32) 3.1.6. Primer Beyin Yaralanmaları Tipleri Primer beyin yaralanmaları tipleri fokal (örn. kafatası kırığı, intrakranyal kanama, laserasyon, kontüzyon, penetre yaralanmalar) ya da diffüz (diffüz aksonal yaralanmalar) olabilir (32). 12 3.1.6.1. Skalp Laserasyonları Skalp laserasyonları travmanın yerini işaret ederler. Yoğun kanayabilirler. Eğer beraberinde çökme kırığı varsa intrakranyal enfeksiyon için giriş yeridir (43). 3.1.6.2. Kafatası Kırıkları Genelde yaralanma ne kadar şiddetli ise kırık olasılığı o kadar artar. Kafatası kırığı olan hastaların kafatası kırığı olmayanlara göre intrakranyal kanama olasılığı daha fazladır. Kafatası kırıkları kubbe veya kaide kırıkları olabilir. Şiddetli kafa yaralanmasında kubbe kırığı olma insidansı % 62 dir. Bunların % 17 si kaideye uzanım gösterir. Kaide ile sınırlı olanların insidansı % 4 dür (43). Şiddetli beyin travmalarına kafatası kırıkları eşlik edebilir. Kanama, kranyal sinir yaralanmalarına yol açabilir ve Kubbe kırıkları çizgisel karakterde olabilir ve sinüslere uzanabilir. Yıldızvari, kapalı veya açık kırık tarzında olabilir. Kapalı kırıklarda dış dünya ile temas yokken açık kırıklarda ise bu temas santral sinir enfeksiyonları için giriş kapısı oluşturur (32). Kırıklar, fragmanların içeri doğru yer değiştirip değiştirmediğine göre deprese ya deprese olmayan kırıklar olarak da tanımlanabilir. Basit kırıkta yalnızca bir kemik fragmanı söz konusu iken bileşik tarz kırıkda en az iki kemik fragmanı vardır. Bazal kırıklar ise genellikle dağılan kuvvetlere bağlı olarak meydana gelir ve beraberinde kranyal sinir yaralanmaları ile otore ve rinore de görülebilir. Rinore, otore ve pnömosefalinin klinik olarak, enfeksiyon açısından önemlidir. Posttravmatik epilepsi olasılığını artırır (43). 3.1.6.3. Travmatik intrakranyal Kanamalar İntrakranyal kanama özellikle kafatası kırığı olan hastalarda sık görülen komplikasyondur. Tablo 3’de görüldüğü gibi sınıflandırılabilir: 13 Travmatik intrakranyal kanama 1-Ekstradural (epidural ) kanama 2-İntradural Kanama a-Subdural Kanama b-İntraserebral kanama c-İntraventriküler kanama d-Subaraknoid kanama (SAK) Tablo:3 Travmatik intrakranyal kanama sınıflaması (43). Epidural kanama tüm yaralanma tiplerinin % 2 sinde görülür. Ölümcül kafa yaralanmaların % 5-15’inde bulunur. Kırıkların % 85’inde epidural kanama vardır. Çocuklarda kırık olmasa da epidural kanama görülebilir. En sık temporal bölgede görülür (43). Epidural kanama kafatasına gelen darbe sonucu kırığa bağlı olarak, sıklıkla, dural arter ya da venlerde ve bazen de diploik venlerde oluşan laserasyonlar sonucu meydana gelir. En sık olarak da orta meningeal arterin yırtığı bu tip bir kanamaya yol açar. Arteriyel kaynaklı bir kanama sonucu oluşan kan birikimi nörolojik tablonun hızla bozulmasına neden olur. (32). Subdural kanamalar şiddetli beyin travmalarında genellikle serebral hemisferleri dural sinüslere bağlayan ve subdural boşluğu birleştiren kortikal ven veya pial arterlerin yaralanmaları ile oluşur. Yaklaşık % 13’nde başka beyin hasarına ilişkin kanıt çok yoktur. Akut, subakut ve kronik olarak tanımlanmaktadır. Mortalite yaklaşık %60-80’dir. (32, 43). İntraserebral kanamalar en sık frontal ve temporal loblarda olmak üzere derin hemisferde de görülebilir. Serebellumda daha az sıklıkla görülürler. Parankim içi kanamalar olup aşırı kortikal kontüzyonun eşlik ettiği daha büyük ve daha derin yerleşimli serebral damar yaralanmalarına sekonder gelişen laserasyon veya kontüzyon sonrası meydana gelir. 14 İntraventriküler kanamalar çok şiddetli travmatik beyin yaralanmalarının sonucunda görülür ve prognozu kötüdür (32, 43). Travmatik subaraknoidal kanama ise subaraknoid aralıktaki yüzeyel mikrodamar kanamalarıdır. Başka bir serebral patoloji eşlik etmiyorsa bu tip bir kanama benign kabul edilebilir. Serebral vazospazmı indükleyebilir. Anjiogram yapılan akut kafa yaralanması olan hastalarda sık görülen vazospazmı indükleyen faktörlerden biri olabilir (43). Ancak kan yıkım ürünleri araknoid villusları bloke ederse kommünike ve hatta mevcut kan 3. ya da 4. ventrikülü tıkarsa da nonkommünike hidrosefali oluşur (32). 3.1.6.4. Kontüzyon ve Laserasyon Beyin yüzeyinin kaidedeki kemik çıkıntıları ile temasının neden olduğu fokal bir beyin hasarıdır. Beyin dokusu içinde peteşiyel kanamalar, sıyrıklar ve ezikler bulunmaktadır, doku ve vasküler sistem yırtılmamıştır. Tanım olarak kontüzyon yüzeyinde pia-araknoid sağlamdır. Laserasyonda ise pia-araknoid yırtılmıştır (43). Frontal poller, orbital girus, silvian fissürlerin üst ve altındaki korteks, temporal poller, temporal lopların lateral ve inferior yüzeylerini etkileyen karakterisitik dağılımı vardır. Daha az sıklıkla serebellar hemisferlerin inferior yüzeyleri etkilenir (43). “Kup” ya da “ kontur-kup” kontüzyonlar ise vasküler harabiyet ile doku harabiyetinin bir birleşimi sonucu oluşur. “Kup” kontüzyon, kafatasına direkt olarak gelen darbeye bağlı bir kuvvetin etkili olduğu bölgede, beynin deforme olup tekrar eski şeklini alması sürecinde meydana gelen negatif basıncın sonucu oluşur. Kontur-kup kontüzyon da benzer düzenek ile oluşmakla beraber darbeye bağlı kuvvetin etkili olduğu bölgenin zıt tarafında meydana gelir (32). Herniasyon kontüzyonları, temporal lob medial kısımlarının tentorium serbest kenarı ile temas ettiği alanlarda veya serebellar tonsillerin foramen magnuma temas ettiği yerde görülür. Frontal ve temporal lobdaki laserasyonlar sıklıkla intraserebral kanama ve subdural kanama ile ilişkilidir (43). 15 3.1.6.5. Diffüz Aksonal Yaralanma Diffüz aksonal yaralanma beynin beyaz cevherinde oluşan geniş kapsamlı, jeneralize harabiyettir. Yüksek hıza bağlı akserelasyon ve deserelasyon esnasında başın lateral hareketlerine bağlı olarak falks ve tentoriyumdaki aşırı esneme bu tip yaralanmaya yol açabilir (42). Diffüz aksonal yaralanma iskemi sonucu da meydana gelebilir. Travmayı takip eden bir kaç gün içerisinde, şişen aksoplazmaya ait amorfbelirgin bir şekle sahip olmayan ve retraksiyon topları olarak adlandırılan beyaz cevher içerisine dağılmış aksonal parçalanmalar, diffüz aksonal yaralanmaların karakteristik özellikleri olarak, primer beyin travmasında görülürler. Bu lezyonların dağılımının sentrum semiovaleden beyin sapına doğru olması, travmayı oluşturan ve giderek artan akselerasyon veya deselerasyonun mekanizmasının belirgin bir göstergesi olarak darbenin etkisinin merkeze doğru yaklaşan karakterde olduğunu düşündürür (32). Travmaya bağlı olarak meydana gelen beyindeki hareket bir aksonal yanıtın meydana çıkmasını tetiklemekte ve birbirini takip eden olaylar başlangıçdan itibaren tam ve en son şeklini alarak değil, aksine 6 saate kadar varan bir süreç içinde oluşmaktadır (65, 69). Bu olaylar aksolemmanın katlanması ve aksoplasmik akımın kesilmesi ve de akson halen canlılığını, devamlılığını korurken herhangi bir devrede aksonda lokalize şişme ile karakterizedir. Sürecin devamı halinde aksondaki ayrılmayı distalde Wallerian dejenerasyonu takip eder. Bu nedenle, travmayı takip eden ilk bir kaç saat içerisinde, en azından, bazen diffüz aksonal yaralanma tablosunu düzeltebilmek mümkün olabilir. Fokal ve polar kontüzyonunu oluşturan süreç, beyin nekrozunu da meydana getirmek üzere saatlerce devam eder (32). Sitotoksik işlemler serbest oksijen radikallerinin özellikle yüksek reaktif hidroksil radikal tarzında salınımı, hücre zarlarının lipid peroksidasyonu, iyon kanallarının kalsiyum akımına açılması, sitokinlerin salınımı ve serbest yağ asitlerinin vazokontrüksiyon ve iskemiye neden olabilen aşırı vazoreaktif maddelere dönüştüren metabolik değişikliklere uğraması şeklinde özetlenebilir (32, 65). 16 Diffüz aksonal yaralanma Gennarelli ve ark. tarafından 3 dereceye ayrılmıştır: İlkinde lezyon, başlıca, serebral hemisferlerin parasagital beyaz cevherinde iken ikincisinde ek olarak korpus kallozumda da görülür. Üçüncü derecede ise bunlara serebral pedinküllerdeki fokal lezyonlar da eşlik eder (37). Diffüz aksonal yaralanmada BBT başlangıçda % 50–80 inde normaldir. Başlangıç karakteristik bulgular korpus kallozum ve beyin sapında gri-beyaz cevher bileşim bölgesinde küçük peteşiyel kanamalardır. Daha sonra çekilen BBT’de ödem veya atrofi gözlenebilir. Yırtılma-ayrılma yaralanmalarının olduğu bölgelere işaret eden küçük, fokal, düşük dansiteli alanlar görülebilir. Serebral hemisferlerde 2 cm’den küçük tek ya da multipl intraparankimal hemorajiler, intraventriküler hemoraji, korpus kallozumda hemoraji, 3. ventrikül komşuluğunda 2 cm’in altında fokal küçük kanama odakları, beyi sapı kanaması ile karakterizedir. Diffüz aksonal yaralanmada en sık gözlenen MRG bulguları ise korpus kallozumun spleniumunda veya beyaz cevherde temporal ya da parietal kortikomedüller kavşakda multifokal anormal sinyaller (T2 ağırlıklı imajlarda parlak) ve dorsolateral rostral midbrain ve korona radiata da dâhil olmak üzere diğer bölgelerde sıklıkla anormal sinyaller ile nonspesifik atrofik değişikliklerdir. (32). 3.1.7. Sekonder Beyin Yaralanmaları 3.1.7.1. Sebepleri Sekonder beyin yaralanmaları primer travmanın etkileriyle daha ileri hücresel harabiyetler sonucunda oluşur. Primer travmayı takiben saatler ya da günler içinde meydana gelir. Hemen, daima, bütünüyle iskemik tipte olup % 80’in üzerinde fatal seyreder (42). Travmatize beyni sekonder olarak etkileyen sistemik olaylar hipoksemi, arteriyel hipotansiyon, hiperkapni, şiddetli hipokapni, ateş, hiponatremi, anemi, diffüz intravasküler koagülopatidir. İntrakranyal olaylar ise kanama 17 (ekstradural, subdural, intraparankimal), beyin şişmesi-ödemi, intrakranyal hipertansiyon, serebral vazospazm, intrakranyal enfeksiyon, epilepsidir. Sekonder beyin harabiyetinde major üç neden olarak hipoksi % 30 (PaO2 < 65 mmHg), hipotansiyon % 13 (sistolik kan basıncı < 90 mmHg ) ve anemi % 12 (hematokrit < % 30 ) bulunmuştur (32). 3.1.7.2. Fizyopatotoloji Travmatik beyin yaralanması sonrasında serebral yaralanamanın ilk evreleri direkt doku hasarı ile serebral kan akımı (SKA) ve metabolizmanının regülasyonunun bozulması ile karakterizedir. Bu “iskemi benzeri” model anaerobik glikolize bağlı laktik asit birikimi, membran permeabilitesinin artması ve sonuçta ödem oluşumuna yol açar. Anaerobik metabolizma hücrenin enerji durumunu devam ettirmesi için yetersiz olduğundan adenozin trifosfat (ATP) depoları boşalır ve enerjiye bağlı membran iyon pompaları bozulur (71). Fizyopatolojik kaskadın ikinci basamağı eksitatör nörotransmitterlerin (glutamat, aspartat) fazla salınımı, N-metil-D-aspartat, α-amino-3hidroksi-5-metil-4-izoksazolpropionat ve voltaja bağımlı Ca+2 ve Na+ kanallarının aktivasyonuyla birlikte terminal membran depolarizasyonu ile karakterizedir. Sonuçta Ca+2 ve Na+ içakımı kendi kendini tüketen (katabolik) intrasellüler süreçlerin başlamasına yol açar. Ca+2 lipit peroksidazlar, proteazlar ve fosfolipazları aktive ederek serbest yağ asitlerinin ve serbest radikallerin intrasellüler konsantrasyonunu arttırır (76). Kaspazlar (ICE benzeri proteinler), translokazlar ve endonükleazların aktivasyonu biyolojik membranlar ve nükleozomal deoksiribonükleik asit (DNA)’da (DNA fragmantasyonu ve DNA tamirinin inhibisyonu) ileri yapısal değişiklikleri başlatır. Bu olaylar birlikte vasküler ve sellüler yapıların membran parçalanmasına ve sonunda programlanmış hücre ölümüne (apoptozis) yol açar (105). Laboratuvar hayvanları ve insanlardaki çalışmalar ile TBY’nin SKA üzerindeki etkilerini araştırılmıştır. TBY sonrası çok erken evrelerden geç evrelere kadar belirli bir zaman aralığında SKA’nı değerlendirmek için sabit xenon BBT ve 133 Xe sintilasyon saptaması, 133 Xe BBT, 15 O2 pozitron emisyon BBT kullanılarak gerçekleştirilen birçok araştırma fokal veya global serebral iskeminin sıklıkla ortaya çıktığını göstermiştir (15, 22, 51, 77). 18 Toplam iskemik beyin hacmi ortalama % 10’dan az olabilir. Serebral iskemi, ölüm veya vejetatif durum ile ilişkilidir (22, 51, 77). Serebral hipoperfüzyonun sıklıkla kötü sonuç ile ilişkili olması TBY ve iskemik inmenin aynı temel mekanizmaları paylaştığını akla getirmektedir. Bu varsayım bir yere kadar doğru olsa da bu iki primer yaralanma tipi arasında ana farklılıklar vardır. Örneğin, geri dönüşümsüz doku hasarının gelişmesi için SKA eşiği TBY’li hastalarda 15 ml/100g/dak iken iskemik inmeli hastalarda 5–8,5 ml/100g/dak’dır (24). Serebral iskemi esas olarak metabolik stres ve iyonik karmaşaya yol açarken, kafa travmasında buna ilaveten beyin dokusu yırtılma güçlerine maruz kalır, nöron hücre gövdeleri, astrositler ve mikrogliada ardışık yapısal yaralanma ve serebral mikrovasküler ve endotel hücre hasarı olur (16, 29, 84). Mekanik yer değiştirme sonucu morfolojik yaralanma (örn. vasküler distorsiyon), otoregülasyonun bozulunca hipotansiyon, nitrik oksit veya kolinerjik nörotransmitterlerin yetersizliği ve prostaglandinin indüklediği vazokonstriksiyonun artması posttravmatik iskeminin mekanizmaların içinde yer alır (3, 29, 105). Travmatik beyin yaralanması olan hastalarda yaralanmanın erken evrelerinde serebral hiperperfüzyon (>55 ml/100g/dak. ) gelişebilir. Benzer şekilde posttravmatik iskemiyi hemen hiperemi izleyebilir. (58, 70) SKA’ndaki artışlar serebral kan hacminde artış ve bunun sonucunda KİB artış ile birlikte vazoparaliziye neden olur. Bu durum metabolik gereksinim ötesinde iskemi kadar zararlı görünmektedir (57). Hem serebral iskemi hem de hiperemi SKA ve serebral metabolizma arasında uyumsuzluk demektir. Örneğin, metabolik hız normal veya yüksekken akımın düşük olması iskemik bir durum olup, metabolik hız normal veya düşükken SKA’nın yüksek olması halinde serebral hiperemi olur. (58, 105). Herhangi bir zamanda yeterli SKA sağlanmasında serebrovasküler otoregülasyon ve CO2- tepkisi önemli mekanizmalardır. Her ikisi de serebral perfüzyon basıncı (SPB) ve KİB’ın devam ettirilmesinin temelidir. Bu düzenleyici mekanizmaların bozulmaları halinde sekonder beyin hasarı riski artar (1). Travmatik beyin yaralanmasından sonra hastaların çoğunda SKA otoregülasyonu bozulmuştur veya ortadan kalkmıştır. SKA otoregülasyonu travmadan 19 hemen sonra ya da daha sonra, geçici veye sürekli olarak bozulabilir. SKA otoregülasyonundaki bozulma hasarın şiddetinden etkilenmez ( 29). Serebrovasküler CO2- tepkisi (yani, hipo- veya hiperkapniye yanıt olarak serebrovasküler konstriksiyon veya dilatasyon) serebral kan akımı otoregülasyonu ile karşılaştırıldığında daha güçlü bir etkidir. Şiddetli beyin yaralanması oaln hastalarda travmadan sonraki erken evrelerde CO2- tepkisi azalmıştır. Hiperemik durumlarda KİB tedavisinin hedefi olarak bu fizyolojik prensibin gösterildiği diğer hastaların çoğunda CO2- tepkisi sağlamdır veya artmıştır (61). Serebral metabolizma (serebral oksijen ve glukoz tüketimi) ve serebral enerji durumu (fosfokreatin ve ATP nin dokudaki konsantrasyonu veya indirekt olarak laktat/piruvat oranı) sıklıkla TBY’den sonra azalır (24, 40). Metabolik bozulmanın hızı primer sebebin şiddetine bağlıdır. Metabolik disfonksiyonu olmayan veya çok az olanlarla karşılaştırıldığında metabolik hızları düşük olan hastalarda sonuçlar daha kötüdür. Travma sonrasında serebral metabolizmada azalma solunum hızının ve ATP üretiminde azalma, mevcut nikotinik koenzim havuzunda ve intramitokondriyal Ca2+ yükünde azalma ile mitokondriyal disfonksiyona yol açan primer nedene bağlıdır (98, 103). Metabolik bozulmayı düzeltme girişimi olarak hiperoksi kullanımı tutarsız sonuçlar ortaya çıkarır (66). Serebral metabolik gereksinimdeki azalmalar SKA’nda azalma ile ilişkili olabilir veya olmayabilir (24). Alternatif bir fizyopatolojik olay olarak glukoz hipermetabolizması olabilir. Bunu geçici de olsa ardışık nöroeksitasyonlar ile birlikte SKA’daki artışların yeterince karşılayamadığı (eş zamanlı) masif transmembran iyon içakımları yönetir. Bu tip akım metabolizmasındaki eşleşmeme sekonder iskemik sebeplerin gelişimini destekler. Travmatik beyin yaralanması beyine gelen oksijen ve serebral oksijen tüketimi arasındaki dengesizlik ile karakterizedir. Bu uyumsuzluğu çok sayıda vasküler ve hemodinamik mekanizma indüklemekteyse de kesin son nokta beyin dokusu hipoksisidir. Travmatik beyin yaralanması olan hastalarda doku hipoksisinin insidansı, süresi ve genişliği sonucun kötü olması ile uyumludur. Beyinde oksijen yoksunluğu ardından sekonder beyin hasarı SPB veya KİB normal olsa bile ortaya çıkabilir. Beyin dokusu oksijen basıncı parametresini KİB veya SPB kılavuzluğundaki tedavi algoritmaları ile 20 birleştiren klinik protokoller oksijen taşınması ve oksijen ihtiyacı arasındaki etkileşim hakkında önemli bilgiler sağlamış, tedavi kişiye göre kritik beyin dokusu oksijenasyonuna dayandırıldığında sonuçların iyileştiğini göstermiştir (54, 97). Travmatik beyin yaralanması iskemik reperfüzyon yaralanması ile benzerlikler gösteren karmaşık bir immünolojik/enflamatuvar doku yanıtı yelpazesine neden olur. Hem primer hem de sekonder nedenler proenflamatuvar sitokinler, prostaglandinler, serbest radikaller ve kompleman dahil olmak üzere hücresel mediyatör salınımını aktive eder. Bu süreçler kemokinleri ve adezyon moleküllerini indükler ve sonrasında immün ve glial hücreleri paralel ve sinerjik olarak harekete geçirir (64, 79). Aktive polimorfonükleer lökositler adezyon moleküllerinin aracılığı ile hem defektif hem de sağlam endotel hücre tabakalarına yapışır. Bu hücreler makrofajlar ve T-hücre lenfositlerinin yanı sıra yaralı dokuya sızarlar (109). Bu enflamatuvar süreçle yanıt olarak yaralı ve komşu doku yok edilir, saatler, günler ve haftalar içinde astrositler mikrofilamantler ve nötrofinler üreterek skar dokusu sentez ederler. Yaralanmadan sonraki saatler içinde tümör nekroz faktörü, interlökin-1-β ve interlökin-6 gibi proenflamatuvar enzimlerin miktarı artar. Doku hasarının ilerlemesi direkt olarak nörotoksik mediyatörlerin salınımına veya indirekt olarak nitrik oksit ve sitokinlerin salınımına bağlıdır. Vazokonstriktörlerin biraz daha salınması (prostaglandinler ve lökotrienler), lökosit ve trombosit adezyonu ile mikrovasküler yapının obliterasyonu ve ödem oluşumu doku perfüzyonunu daha da azaltır ve ardından sekonder beyin hasarını şiddetlendirir (105). Travmatik beyin yaralanmasından sonra iki farklı hücre ölümü tipi görülebilir: nekroz ve apoptoz (programlanmış hücre ölümü). Nekroz, eksitatör amino asid nörotransmitterlerin aşırı salınımı ile birlikte şiddetli mekanik veya iskemik/hipoksik doku hasarına ve metabolik yetmezliğe yanıt olarak ortaya çıkar. Sonrasında fosfolipazlar, proteazlar ve lipid peroksidazlar biyolojik membranları otolize uğratır. Sonuçta ortaya çıkan hücre artığı ‘antijen’ olarak algılanır ve enflamatuvar süreçler tarafından arkada skar dokusu kalacak şekilde ortadan kaldırılır. Apoptoza uğrayan hücreler travmadan hemen sonraki dönemde fizyolojik membran potansiyeli sağlayan yeterli ATP üretimi ile morfolojik olarak sağlamdırlar. Apoptoz primer nedenden saatler veya günler sonra görülebilir hale gelir. Fosfatidilserin 21 translokasyonu nükler membranların parçalanması, kromatin kondansasyonu ve DNA parçalanması ile birlikte membran bütünlüğünün farklı, ilerleyici şekilde bozulmasını başlatır. Yoğunlaşmış intrasellüler materyalden türeyen çok küçük partiküller (apoptotik cisimler) eksitotoksik mekanizmalar ile küçülmekte olan hücreden temizlenir. Apoptozun doğası için genelde enerji kaynağına ve doğal olarak var olan pro- ve antiapoptotik proteinler arasında dengesizliğe ihtiyaç vardır. Programlanmış hücre ölümünün en önemli mediyatörlerinin interlökin dönüştürücü enzim ailesinin spesifik proteazlarını temsil eden kaspazların ardışık aktivasyon ve deaktivasyonu olduğu saptanmıştır. (31). Apoptozun klinik anlamlılığı hücresel yıkımın geç başlangıcına, terapötik girişimler açısından da gerçekçi fırsat pencerelerinin açılması imkânını sunulmasına bağlıdır. 3.2. Beyin Ödemi Travma, beyin tümörü, toksik, anoksik, metabolik ve enfeksiyon kaynaklı olabilen beyin ödemi, morbidite ve mortaliteyi olumsuz yönde etkileyen bir durumdur. Beynin sıvı hacmindeki bu artış, kafa içi basıncı artırmanın yanı sıra perfüzyon ve oksijenasyonun bozulmasına neden olur. Diğer organlardan farklı olarak beyin, su içeriğinin artışına çok daha duyarlıdır ve beyin ödemi yaşamı tehdit eden bir durumdur (59). 3.2.1. Ödemin sınıflandırılması Beyin ödemi sınıflandırması ilk kez 1967’de yapılmıştır. Buna göre vazojenik ve sitotoksik olmak üzere iki tip ödem mevcuttur. 1975’de bu sınıflamaya hidrosefalik hastalarda gözlenen interstisyel ödem ve osmotik dengenin bozulmasından kaynaklanan ozmotik ödem de eklenmiştir (102). Ozmotik ödemde özellikle sodyum (Na+) gibi elektrolitlerin dengesindeki bozulma ve hücre içine su girişindeki artış söz konusudur. Hidrosefalik/ interstisyel ödemde beyin-omurilik sıvısının akışında sorun vardır. Vazojenik ve sitotoksik ödem oluşumunda ise birçok faktörün rolü vardır. Bunlar arasında glutamat, NO, araşidonik asit ve metabolitleri, histamin, kininler, interlökinler, vasküler endotelyal büyüme faktörü (VEGF), laktat, serbest oksijen radikalleri, potasyum (K+), hidrojen (H+) ve kalsiyum (Ca2+) sayılabilir. Ödem sınıflandırması ve bu ödem tiplerinin özellikleri, oluşum mekanizmaları Tablo 4’de özetlenmiştir (100). 22 Neden Vazojenik Sitotoksik KBB hasarı, İskemi, Anoksi, Akut: travma, İntoksikasyon inme, hemoraji, arteriyel hipertansiyon Kronik: Tümör, apse, ensefalit Ozmotik Metabolik bozukluk: Diyaliz, Dehidratasyon, Diyabetik koma Interstisyel Obstriktif hidrosefali Oluşumu Doku hasarı sonucu kapiller endotel hücrelerinin geçirgenlik artışı 1. Hücre Ozmotik zarında gradient Na/K (plazma→doku) geçirgenliğinde artış 2. Na/K-ATPaz pompasında bozukluk 3. Osmotik maddelerin alımında artma Beyinomurilik sıvısının geri emilimi ya da efluksunun bozulması Geçirgenlik Ödem sıvısının özelliği Artar Proteinden zengin Değişmez Protein yok Elektrolitten zengin Değişmez Protein yok Elektrolit oranı çok yüksek veya çok düşük Değişmez Normal beyin omurilik sıvısı Morfoloji Interstisyel boşlukta genişleme, hücreler şişmez; çoğunlukla beyaz madde etkilenir, sekonder olarak astrositlerde şişme gözlenir. Hücreler şişer; interstisyel boşluk azalır Hücreler şişer; interstisyel boşluk azalır Sıvı hücreler arasıdır; Interstisyel boşlukta genişleme Tablo:4 Ödem sınıflandırması (59, 100, 102) 23 Son TBY çalışmalarının sonuçları bu yaralanmalarda hakim olan ödem tipinin hücresel ödem olduğunu göstermiştir. Bu sonuçları başka araştırmalar doğrulamıştır (102). Travmatik beyin yaralanmasında iyonik disfonksiyon olduğu ve mekanik zarar ile eş zamanlı olarak ortaya çıkan depolarizasyon sonucunda ekstrasellüler K+’un geçici süre ile arttığı kanıtlanmıştır. Bu iyonik homeostaz kaybına beraberinde sodyum hareketi eşlik etmelidir (102). Travmatik beyin yaralanmasının mekanik deformasyon, nörotransmitter salınımı mitokondri disfonksiyonu ve membran depolarizasyonu normal iyon gradiyentlerinde değişikliğe neden olabilecek olaylar kaskadını tetiklediği düşünülmektedir. Mekanik deformasyon ile eksitatuvar amino asidler salınır ve membran depolarizasyonu iyonların elektrokimyasal gradiyentlerinden aşağıya doğru hareketine izin veren ligand kapılı iyon kanallarını aktive edebilir. İyon akımı ve travma sonucu membran depolarizasyonu voltaja duyarlı iyon kanallarını tetikleyebilir, sonrasında iyon hareketine başka yollar sağlar. Bu iyonik bozukluklar ekstrasellüler K+’da artış, beraberinde ekstrasellüler Na+, Ca+2 ve Cl-‘da azalma ile belirlenir. İyonik homeostazın yeniden sağlanması Na+-K+ adenozin trifosfataz, Na+/K+/2Cl- cotransporter, Na+-H+ transporter Na+-Ca+2 değiştiricisi gibi enerjiye bağlı co- ve counter (birlikte ve karşılıklı) işlemler veya vasküler ya da BOS kompartmanına basit sürüklenme yolu ile olabilir. Beyin yaralanmasına eşlik eden iyonik hareketteki net dengenin ekstrasellüler boşluklardan hücrelerin içine katyon hareketine neden olduğundan kuşkulanmak mantıklıdır. Na+ ve Ca+2 hareketini elektronötraliteyi korumak için pasif olarak Cl- ve bunu da izoozmotik olarak su takip eder. Devam ederse bu iyonik bozukluklar KİB artışının esas katılımcısı olduğu gösterilen hücre şişmesi ve sitoksik ödeme neden olabilir (68). 3.3. Kan Beyin Bariyeri Kan beyin bariyeri (KBB) beyindeki vasküler endotel hücrelerinin oluşturduğu bir yapıdır. Endotel, astroglia, perisit hücreleri ve bazal laminanın oluşturduğu bu sistem dolaşımdaki maddelerin beyne geçmesini kısıtlar. Astrositlerin glial ayakları ve perisit adı verilen ve endotel hücrelerinin büyümesini kontrol eden kontraktil hücreler bazal laminaya tutunan serebral endotel hücrelerini sıkıca sararlar. Bunlarla birlikte sıkı 24 bağlantıları olan tek sıra hücreler KBB’ni oluşturmaktadır. Pinositik veziküllerin eksikliği ve pencere bulunmaması da geçişi kısıtlamaktadır (27), Sıkı bağlantılar paraselüler geçişi kısıtlar. Ancak lipofil bileşikler çözünerek ya da difüzyonla geçebilir. Polar bileşikler ise özel taşıyıcılarla taşınır. KBB’nde 20’den fazla spesifik taşıyıcı pompanın varlığı saptanmıştır (95). Beynin yaklaşık % 1’inde KBB’nin bulunmadığı bazı bölgeler vardır. Sirkümventriküler organlarda (medyan eminens, area postrema, subfornikal organ, subkomissural organ, hipofiz bezi, nörohipofiz ve organum vasculosum) KBB yoktur (95). 3.4. Travma sonrası Oksidan Hasar ve Savunma Mekanizmaları Travmatik beyin yaralanması primer ve sekonder olarak özellikle glutamat olmak üzere eksitatör amino asit nörotransmitterlerin masif salınımı ile ilişkilidir (17, 83). Ekstrasellüler glutamattaki bu fazlalık nöronları ve astrositleri etkiler, iyonotropik ve metabotropik glutamat reseptörlerinin aşırı uyarımının ardından Ca+2, Na+ ve K+ akımlarına neden olur. Bu olaylar kan beyin bariyerinin yıkılması, Na+ / K+ - ATPaz aktivitesini ve sonrasında metabolik ihtiyacı arttıran iyonik gradiyentleri kompanse etmeye yönelik hücre girişimi dahil katabolik süreçleri tetiklese de hücre ile eşleşmeyen kötü akım-metabolizması döngüsü oluştururlar. (29, 75). Oksidatif stres travmatik beyin yaralanmasına yanıt olarak reaktif oksijen türlerinin (oksijen serbest radikalleri ve süperoksidler, hidrojen peroksit, nitrik oksit ve peroksinitrit dahil ilişkili varlıklar) oluşumu ile bağlantılıdır. Eksitotoksisite ve endojen antioksidan sistemin (süperoksit dismutaz, glutatyon peroksidaz ve katalaz) tükenmesine bağlı olarak reaktif oksijen türlerinin aşırı üretimi hücresel ve vasküler yapıların peroksidasyonu, protein oksidasyonu, DNA parçalanması ve mitokondriyal elektron transport zincirinin inhibisyonunu indükler. Bu mekanizmaların hemen hücre ölümüne katılımı yeterli ise de oksidatif stres enflamatuvar süreçler ile erken veya geç apoptotik programları indükler (7, 20, 92). 25 3.4.1. Serbest radikaller ve Oksidan hasar “Serbest radikal”, eşlenmemiş elektron içeren atom veya moleküldür. Genelde elektronlar atom veya molekülde eşlenik olarak bulunmaları nedeniyle molekül stabildir ve reaktif değildir. Ancak moleküle bir elektron ilavesi ya da bir elektron kaybı onu kararsız ve reaktif hale getirir. Tablo 5: Başlıca reaktif oksijen metabolitleri ve yarılanma ömürleri Radikal Sembol 37 ºC de yarılanma ömrü (saniye) Alkoksil radikali Hidroksil radikali Lipit peroksid Moleküler oksijen Peroksil radikali Singlet oksijen Süperoksit anyon radikali RO • OH• ROOH O2 ROO• 1 O2 O2− • 1x10-6 1x10-9 >102 >10 1x10-2 1x10-6 1x10-6 Organizmanın aldığı O2’in % 95’i enerji oluşumunda kullanılırken, % 5’den fazlası da reaktif oksijen metabolitlerine, yani bir başka deyişle oksijen türevi radikallere dönüşür. O2-•, oksijen (O2) molekülüne bir elektron ilavesi ile oluşur ve serbest radikal hasarına karşı koruyucu antioksidan bir enzim olan ve oksidan hasar oluşumu ile birlikte artan süperoksit dismutaz (SOD) aracılığı ile H2O2’ye indirgenir. eO2 -------------- ► O2SOD 2 O2-• + 2H+ ------------------------ ► H2O2 + O2 H2O2 eşlenmemiş elektron içermediği için tek başına radikal değildir. H2O2’in metabolizması hücre içinde birkaç şekilde olabilir: 1- H2O2, katalaz veya glutatyon peroksidaz (GSH-Px) tarafından toksik olmayan ürünlere (su ve indirgenmiş glutatyon, GSSG) dönüşür; o da NADPH (nikotinamidadenindinükleotidfosfat) ile birleşerek glutatyon (GSH) oluşturur. 26 katalaz a) 2 H2O2 ----------------------- ►2 H2O + O2 GSH-Px b) H2O2 + 2 GSH ------------------------------- ► 2 H2O + GSSG GSH-redüktaz GSSG + NADPH + H+ -------------------------------- ► NADP+ + 2 GSH 2- H2O2 geçiş metallerinin varlığında toksik •OH radikaline dönüşür.(Fenton reaksiyonu) Fe+2-----------------►Fe+3 H2O2 --------------------------------------------------------------------------------------------------------- ► • OH H2O2’nin •OH’e indirgenmesi Fe+2 ve Cu+2 gibi geçiş metalleri varlığında olur. Bu metaller hücrede serbest değildir. Serumda demir transferin, bakır ise seruloplazmin şeklinde depolanır ki; depo halinde iken serbest radikal oluşturamaz. Dolayısı ile antioksidan enzimler ve serbest radikal süpürücülerinin yanı sıra geçiş metallerini bağlayan proteinler de organizmanın total antioksidan kapasitesinin bir bölümünü oluşturur (106). • OH ise oldukça reaktif ve toksik bir radikaldir. Büyük molekül yapısı ve elektronegativitesi nedeni ile DNA, protein, karbonhidrat ve lipidler gibi makromoleküllerle reaksiyona girerek bu yapılarda oksidatif hasara neden olur. Makromoleküller hücrelerde kısıtlı miktarlarda bulunduklarından bu yapılarda oluşan hasar oldukça önemlidir (36). O2-•’e bağlı olan toksisite •OH’e bağlı olanla karşılaştırıldığında daha hafiftir. Ancak bazen radikaller diğer radikallerle etkileşerek stabil moleküller verir. Böyle bir durumda her radikaldeki eşlenmemiş elektronlar kovalan bağ oluşturur. Örneğin, O2-• endojen NO ile tepkimeye girerek peroksinitrit (ONOO-) oluşturur. Oluşan ONOO- protein ve peptitlerin metiyonin rezidülerini ve tiyol gruplarını oksitleyerek hasar oluşturabilir ve ayrıca NO2•, OH• ya da nitronyum iyonu (NO2+) gibi toksik ürünlere dönüşebilir (100). O2- • + NO------------------------ ► ONOO- 27 “Oksidatif hasar” organizmada oluşan toksik oksijen türevi metabolitlerin neden olduğu hücre, doku ve organ hasarını tanımlamaktadır. Oksidatif hasarın göstergeleri olarak kullanılan başlıca parametreler radikallerin hedef aldığı makromoleküllerdeki hasarı yansıtırlar (36). Bu parametreler başlıca; • 8-okso,dihydro-2-deoksikuanosin tayini (DNA hasarı) • Malondialdehit (MDA), hidroksialkenal ve dien konjugatlarının tayini (Lipidlerde oksidasyon) • Karbonil tayini (proteinlerin oksidatif hasarı) dir. 3.4.1.1. Serbest Radikallerin Fizyopatolojik Olaylardaki Rolü Serbest oksijen radikalleri fizyolojik şartlarda endojen olarak oluşurken aynı zamanda hava kirliliği, pestisitler, UV radyasyon, X-ışını gibi ekzojen faktörlerin etkisi ile de oluşumları artmaktadır (36). Serbest radikallerin artrit, hemorajik şok, atheroskleroz, yaşlanma, iskemi/ reperfüzyon hasarı, enfeksiyonlar, sepsis, Alzheimer ve Parkinson hastalığı, gastrointestinal disfonksiyonlar, tümör oluşumu, karsinogenez ve AIDS gibi hastalıklarda işe karıştığı gösterilmiştir (20). Güçlü serbest radikal süpürücü etkiye sahip olan çok sayıda sentetik ve doğal antioksidanın insan sağlığında ve hastalıkların önlenmesinde faydalı etkilerinin olduğu bilinmekte ve bundan yola çıkılarak reaktif oksijen metabolitleri (ROM)’ne bağlı gelişen pek çok hastalıkta kullanılabileceği bildirilmektedir (100). Bu konuda özellikle son yıllarda çalışılmasına rağmen, özellikle nörolojik bozukluklarda kullanılmak üzere KBB’ni geçebilen antioksidanlara ihtiyaç vardır (39). Serbest radikaller yüksek oranda reaktif bileşiklerdir. Sahip oldukları bu reaktivite ile hücre membranın yapısında bulunan çoklu doymamış yağ asitleri, DNA’daki nükleotitler ve proteinlerin sülfidril grupları ile reaksiyona girerek doku hasarına neden olurlar. (25, 26). Poli(ADP-riboz) sentaz (PARS) ya da diğer adıyla poli(ADP-riboz) tranferaz olarak da bilinen poli(ADP-riboz) polimeraz–1 (PARP–1) enzimi ökaryotlarda bulunan nükleer bir enzimdir. Çevresel uyarılar, hidroksil, süperoksit ve peroksinitrit gibi serbest radikaller, iyonizan radyasyon ve genotoksik ajanların PARS enzimini aktive etmesi DNA sarmalında kırılmaya neden olur. PARS enziminin inhibisyonu birçok enflamatuvar hastalıkta koruyucu etki göstermektedir (25, 26). 28 3.4.2. Endojen Antioksidan Sistem Serbest oksijen radikallerinin ciddi hasar yapma potansiyellerine karşı hücreler, bu toksik ürünleri hızla metabolize edecek savunma mekanizmaları geliştirmişlerdir. Hücre ve dokuları serbest oksijen radikallerine karşı korumada oksijen radikallerinin yapımlarının inhibisyonu önemlidir. SOD enzimi O2-"in H2O2’ye dönüşümünü katalize ederek oksijen toksisitesine karşı ilk savunma basamağını oluşturur. Sitozolik SOD bakır-çinko içeren metalloenzimdir ve mitokondride bulunan formu ise manganez içerir. Süperoksitin H2O2 ve moleküler oksijene dönüşümünü katalize eder. Peroksizomlarda bulunan H2O2 yine aynı organelde bulunan katalaz enzimi tarafından metabolize edilir. Katalaz, sitoplazma içinde ve peroksizomlarda ve hemen her hücrede bulunan enzimdir. Sitozolde ve mitokondrilerde bulunan H2O2 metabolizmasında rol alan bir diğer enzim GSH-Px’dır. Organizmada bulunan tüm bu antioksidan enzimler endojen veya hastalık durumunda oluşumu artan serbest radikallere karşı vücudun antioksidan savunma sisteminin önemli bir kısmını oluşturmaktadırlar. Bir diğer önemli antioksidan savunma yöntemi ise Fenton reaksiyonu ile hidroksil radikali oluşumunun engellenerek, redoks aktif metallerin (bakır, demir gibi) transport veya depo proteinlerine bağlanarak ortamdan uzaklaştırılması ve serbest radikallerin lipidlerle olusturduğu zincirleme reaksiyonların durdurulmasıdır. Farklı bir koruma mekanizması da antioksidan etkili vitaminler ile serbest oksijen radikallerinin detoksifiye edilmesidir. E vitamini lipidden zengin membranlarda, C vitamini su bazlı ortamlarda temel korumayı oluşturur. E vitamini (α-tokoferol) lipid peroksidasyonunda oluşan zincirleme reaksiyonu, yapısındaki hidrojen atomunu lipid peroksiline vererek önler. Kendisi ise oksidasyon sonucu α-tokoferoksil radikaline dönüsür. C vitamini (askorbik asit) ise bir elektron vererek dehidroaskorbat’a indirgenirken tokoferoksil radikalinin α-tokoferole dönüşümünü gerçekleştirir. Diğer antioksidan maddeler α-lipoik asit, ürat, γ-glutamil-sisteinil-glisin (glutatyon, GSH) gibi hidrofilik radikal süpürücüler, flavonoidler, karotenoidler, ubiquinol gibi lipid radikal süpürücüler ve GSH redüktaz, dehidroaskorbate redüktaz gibi küçük moleküler antioksidanların okside formlarının indirgenmesinde etkili enzimler, protein tiyollerin devamlılığından sorumlu olan tiyoredoksin redüktaz, GSSG’un GSH’a dönüşümünde elektron donörü olarak kullandığı NADPH yenileyen glukoz–6 fosfat dehidrojenez (G6PD) enzimi gibi enzimler antioksidan savunmaya katkıda bulunmaktadır. 29 Beyin KBB ile korunmuş olmakla birlikte oksidan hasara karşı çok daha duyarlıdır. Bunda endojen antioksidan kapasitenin azlığı ve beynin yağ asidi, askorbat ve demirden zengin olması gibi yapısal faktörlerin rolü vardır (20). 3.5. Sisteinil Lökotrienler Lökotrienler araşidonik asitten türeyen kuvvetli lipit mediyatörlerdir. Proenflamatuvar etkileri vardır. Çeşitli immün ve enflamatuvar uyaranlara yanıt olarak sentez edilirler. Lökotrienlerin üretimi nötrofiller, eozinofiller, makrofajlar ve mast hücreleri gibi enflamatuvar hücrelerin nükleer membranlarında başlar, burada araşidonik asit 5lipooksijenaz enzimi ile araşidonik asit gliserofosfolipitlerden ayrılır. 5-lipooksijenaz araşidonik asidin unstabil LTA4’e dönüşümünü aksesuvar protein 5-lipooksijenazı aktive eden protein (FLAP) yardımı ile katalize eder. LTA4, LTA4 hidrolaz etkisi ile LTB4’e metabolize olur veya glutatyon ile konjuge olarak sistein içeren LT’lere (sisteinil lökotrienlere, yani LTC4, LTD4 ve LTE4’e) dönüşür. LTC4 sentezinin alternatif bir yolu da transsellüler metabolizme olarak bilinen hücreden hücreye kooperasyon yoludur (6, 18). Burada, vasküler endotel hücreleri ve trombositlerde, LTA4 üretimi için enzimatik bir mekanizma yoktur. Nötrofilden türeyen LTA4’den hücresel ortamda LTC4 üretilmesi bir avantaj olabilir. Şekil 2: Lökotrienlerin üretimin şematik gösterilmesi (18) 30 Sisteinil lökotrienlere yanıt veren iki reseptör kopyalanmıştır, CysLT1 ve CysLT2 olarak adlandırılırlar (21, 33). Her ikisi de klasik G-proteinine bağlı reseptörlerdir, yedi transmembran bölgeleri vardır. Sisteinil LTler G-proteini ile eşleşmiş spesifik reseptörleri (GPCRler) aktive ederek biyolojik etkilerini gösterirler. CysLT reseptörleri lökositler ve dalakta mevcuttur. Diğer organlarda varlıkları özelliklidir. CysLT1 reseptörü solunum ve gastrointestinal sistemlerde mevcut iken CysLT2 reseptörü esas olarak kardiyovasküler sistem ve beyin dokusunda bulunur. (18, 108) Northern blot ile tespit edildiği üzere CysLT1 dalak ve periferik kan lökositlerinde büyük miktarda varken, beyin, akciğer, kolon, kalp, pankreas, prostat, iskelet kası, böbrek, karaciğer, plasenta ve ince bağırsakta azdır (108). Sisteinil lökotrienler LTC4, LTD4 ve LTE4 periferik organlarda ve merkezi sinir sistemindeki çeşitli hastalıklarda yer alan güçlü enflamatuvar mediyatörlerdir. CysLT ler düz kas kontraksiyon unu, mikrovasküler sızıntıyı, eozinofillerin olay yerine gelmesini, astım ve rinit gibi enflamatuvar hastalıklarda diğer yanıtları başlatırlar. SSS’de serebral iskemi, beyin travması ve tümörleri dâhil beyin hasralarında CysLT lerin düzeyleri yükselir. Sıçan modelinde CysLT lerin beyin travması sonrasında yükseldiği, 4. saat ve 7. günde pik yaptığı, bunun da ödeme ve hücresel enflamatuvar yanıta bağlı olduğu bildirilmiştir (108). Akut enflamasyonun özelliği olarak dokuya PMN lokösit infiltrasyonu kemotaktik mediatörlerin kolektif etkisinin var olduğunu işaret eder. Çeşitli kemokinler ve bir lipit mediatör metoboliti olan CysLT’ler iskemide güçlü inflamatör mediatörlerdir. Nötrofiller iskemik alana göç ettiğinde reaktif oksijen ürünleri, proteazlar, elastazlar, MPO, sitokinler ve doku zedelenmesinde yer alan çeşitli diğer sitokinleri salarlar. Montelukast eozinolotik enflamasyonu bu hücrelerden enflamatuvar sitokin salınımını inhibe eder (107). LTD4’ün bağlanması montelukast gibi yapısal olarak farklı CysLT1 antagonist sınıfları tarafından kuvvetle ve yarışmalı olarak inhibe edilir (85). Beyinde varlıklarının az olduğu bildirilmişse (18) de TBY veya tümörün olduğu insan beynin de oldukları daha sonra kanıtlanmıştır (108). CysLT2’nin mRNA’sı özellikle hipotalamus, talamus, putamen, hipofiz ve medullada olmak üzere beynin çeşitli bölgelerinde oldukça fazla miktarda mevcuttur (85). Beyin parenkiminde granülositler ile hem travmatik yaralanmanın geç evresinde hem de tümörleri çevreleyen alanda, TBY sonrasında rejenere olan mikrodamarların endotel hücrelerinde mevcut oldukları bildirilmiştir (48). 31 3.6. Montelukast Sodyum MK-0476 (1-(((1(R)-(3-(2-(7-chloro-2-quinolinyl)-(E)-ethenyl)phenyl)(3-2-1(1-hydroxy1-methylethy)phenyl)propyl)thio)methyl)cyclopropane asetik asit sodyum tuzu olarak da bilinir. Montelukast sodyum, LTD4 ün kuvvetli ve selektif bir inhibitörüdür. Biyokimyasal ve farmakolojik biyoanalizlere dayanarak CysLT1 reseptörüne yüksek bir afinite ve seçicilikle bağlanır (55). Montelukast, herhangi bir agonistlik aktivite göstermeksizin CysLT1 reseptörüne LTC4, LTD4 ve LTE4’ün fizyolojik etkilerini kuvvetle inhibe eder (55). Şekil:3 Montelukast Sodyum’un moleküler yapısı (10) 3.6.1. Farmakokinetik Özellikleri Oral uygulamadan sonra montelukast hızla ve hemen hemen tümüyle emilir. Doruk plazma konsatrasyonuna (Cmax) 3 saatte ulaşır. Montelukast plazma proteinlerine % 99’dan fazla bağlanır. Montelukast karalı durum dağılım hacmi ortalama 8-11 litredir. Montelukast büyük oranda metabolize olur. Montelukastın metabolizmasında sitokrom P450 3A4 ve 2C9 un rolü olduğu gösterilmiştir. Montelukastın plazma klerensi ortalama 45 ml/dk’dır. Montelukastın metobolitlerinin hemen hemen tümü safra ile atılmaktadır. Montelukastın ortalama plazma yarılanma ömrünün 2,7–5,5 saat arasında değiştiği gösterilmiştir (6). Oral uygulamadan sonra montelukast hızla ve hemen hemen tümüyle emilir. Ortalama oral biyoyararlanım, %64’tür. Tedavi dozlarıyla yapılan çalışmalarda, erişkin ve çocuk hastalarda montelukastın metabolitlerinin plazma konsantrasyonları kararlı durumda saptanamaz. 32 3.6.2. Farmakolojik Etkileri Montelukast sodyum sisteinil lökotrien CysLT1 reseptörünü spesifik olarak inhibe eden, selektif ve oral yoldan etkin bir lökotrien reseptör antagonistidir. Sisteinil lökotrienler ( LTC , LTD , LTE ) mast hücresi ve eozinofiller dâhil çeşitli 4 4 4 hücrelerden salınan güçlü enflamatuar eikozanoidlerdir. Bu önemli pro-astmatik mediatörler, insanın solunum yolunda bulunan CysLT reseptörlerine bağlanır. CysLT1 reseptörü, solunum yolu düz kas hücreleri ve solunum yolu makrofajları da dâhil olmak üzere insan solunum yolunda ve diğer pro-enflamatuar hücrelerde (eozinofiller ve belirli miyeloid kök hücreler dâhil) bulunur. Astım ve alerjik rinitin patofizyolojisiyle, CysLT’ler arasında ilişki vardır. Astımda lökotrien aracılı etkiler; bronkokonstriksiyon, müköz sekresyon, damar geçirgenliği ve eozinofil birikimi gibi bir dizi solunum yolu aktivitesini içerir. Alerjik rinitte, CysLT’ler hem erken hem de geç fazlı reaksiyonlar sırasında alerjene maruz kaldıktan sonra burun mukozasından salgılanır ve alerjik rinitin semptomlarıyla ilişkilidir. CysLT’lerin intranazal yoldan uygulanması sonucu, nazal solunum yolu direncinin ve nazal tıkanma semptomlarının arttığı gösterilmiştir. Montelukast, astıma bağlı enflamasyon parametrelerini anlamlı derecede iyileştiren, güçlü oral yoldan etkin bir bileşiktir. Biyokimyasal ve farmakolojik biyoanalizlere dayanarak CysLT1 reseptörüne (prostanoid, kolinerjik veya β-adrenejik reseptör gibi diğer farmakolojik açıdan önemli havayolu reseptörleri yerine) yüksek bir afinite ve seçicilikle bağlanır. Montelukast herhangi bir agonist aktivite göstermeksizin CysLT1 reseptöründe LTC , LTD , 4 4 LTE 'ün fizyolojik etkilerini kuvvetle inhibe eder (6). 4 Sisteinil LTler astımın akut ve kronik belirtileri ile alerjik rinit (AR) ve diğer nasal hastalıkların fizyopatolojisinde önemli rol oynarlar. Sisteinil LT’lerin astım ve AR’in klinik belirtilerindeki önemi Lökotrien reseptör antagonistlerinin tedavideki faydaları gösterilerek anlaşılmıştır. Kolon kanseri, atopik dermatit (AD) ve ürtiker dâhil diğer enflamatuvar durumların fizyopatoljisinde sisteinil LT lerin oynadığı rol üzerine yazılan güncel yazılar LT reseptör antagonistlerinin bu hastalıkların tedavisinde potansiyel etkisi olduğuna dikkat çekilmesi için yeterlidir (18). İskemi, travma, tümör, MS, ensefalomyelit ve yaşlanma gibi çeşitli SSS hastalıklarında yer alan güçlü enflamatuvar mediatörlerdir. İskemik beyinlerde CysLT’lerin üretimi artar. Artan CysLT’ler kan-beyin bariyer disfonksiyonu ve beyin ödemi ile 33 uyumludur. Bu nedenle CysLT’lerin beyinde birikimi serebral iskemi anahtar rol oynayabilir (107). Sisteinil LT1 reseptör antagonisti olan montelukastın, iskemik reperfüzyonun indüklediği nötrofil birikmesini, oksidatif hasarı ve renal disfonksiyonunu azalttığını kesin olarak gösterilmiştir. Montelukastın reperfüzyonun neden olduğu hasarı önleyici bu etkileri en azından nötrofil infiltrasyonunu inhibe etmesine, oksidan-antioksidan durumu dengelemesine ve enflamatuvar mediyatör oluşumunu düzenlemesine bağlanabilir (90). Spinal kord yaralanması modelinde nöroproteksiyon etki yaptığını gösterilmişdir. (38) 34 4. Gereç ve Yöntem Bu çalışma S.B. Haydaypaşa Numune Eğitim ve Araştırma Hastanesi Deney Hayvanı Etik Kurulu aldıktan sonra yine aynı hastanenin Deneysel Araştırma ve Hayvan Laboratuarı ve Marmara Üniversitesi Eczacılık Fakültesi Farmakoloji Laboratuarında gerçekleştirilmiştir. 4.1. Deney Hayvanları Bu çalışmada S.B. Haydaypaşa Numune Eğitim ve Araştırma Hastanesi Deneysel Araştırma Laboraturarından temin edilen 64 adet ortalama ağırlıkları 300 g olan Spraque- Dawley soyu erişkin erkek sıçanlar kullanıldı. Deneyler Marmara Üniversitesi Eczacılık Fakültesi Farmakoloji A.B.D. laboratuarlarında gerçekleştirilmiştir. 4.2. Kafa Travması Modeli Kafa travması modeli oluşturmak üzere Marmarou’nun impakt akselerasyon modeli modifiye edilerek kullanıldı (35, 67, 101). Bu modelde iç çapı 18 mm ve dış çapı 19 mm olan silindir tüp kullanıldı (Resim 1). Resim 1: Kafa travmasın oluşturulmasında kullaılan düzenek 35 Kafa travması oluşturmak üzere cerrahi girişim yapılacak ratlara ketamin ve klorpromazin perkütan intraperitoneal (ip) verildi ve ratlar mantar bloklar üzerinde prone pozisyonda yerleştirildi (Resim 2). Resim 2: Sıçanların travmaya hazırlanması Kafatasında oluşacak çökme kırıklarını önlemesi açısından koronal ve lamdoid sütür arası cilt insizyonu takiben 10 mm çapında 3mm kalınlığında çelik disk konuldu (Resim 3). Resim 3: Çelik disk yerleştirilmesi 36 Bir metre yükseklikten 300 g ağırlığındaki kitle, serbest düşüş yapacak şekilde düşürülerek kafa travması oluşturuldu. Kafa travması oluşturulan 3. grup ratlara 10 mg/kg intraperitoneal montelukast verildi. Hemostazı takiben tabakalar usulüne uygun olarak kapatıldı. Ratlar normal oda ısısında normal olarak uyandırıldı. Uygun ortam ısısının sağlandığı ısıtılmış bölümde herbiri ayrı kafeste olmak üzere standart fare yemi ile beslendi. Sıçanlar 4 gruba ayrıldı: Grup 1 (n:8 rat) Kontrol grubu Grup 2 (n:8 rat) Montelukast kontrolü Grup 3 (n:8 rat ) Kafa travması uygulandı Grup 4 (n:8 rat) Kafa travması uygulandı ve 10 mg/kg intraperitoneal montelukast verildi. 4.3. Nörolojik Muayene Hayvanlara travma oluşumunu takip eden 48. saatte nörolojik muayene yapılmıştır. Nörolojik muayene Toklu ve arkadaşlarının oluşturduğu modifiye Bederson nörolojik skorlaması ile yapılmıştır (9, 100). Bu skorlamada bilinç durumları, tırmanma yetenekleri, ekstremite tonusları ve ağrılı uyarana yanıtları değerlendirilmiştir. Puanlama sisteminde düşük skor (asgari 0) hayvanın uyanık, aktif ve reflekslerinin normal olduğunu göstermektedir. Artan skorlar (azami 20) nörolojik bozukluğun meydana geldiğine işaret etmektedir. Ayrıntılı puanlama sistemi Tablo 6’de gösterilmiştir. 37 Normal 0 Huzursuz 1 Letarji 2 Stupor 3 Nöbet 4 Koma 5 Tırmanıyor 0 TIRMANMA Arka bacak üzerinde duruyor 1 PLATFORMU >5s asılı kalıyor 2 <5s asılı kalıyor 3 Yakalama refleksi yok 4 EKSTREMİTE Normal 0 TONUSU Zayıf 1 YÜRÜME Normal 0 Hipomobilite 1 Pençe addüksiyon 2 Paretik tarafa dönme 3 Ayakta duramıyor 4 >1s 180 derece 0 <1s 180 derece 1 1-3s 90 derece 2 <1s 90 derece 3 1-3s 45 derece 4 <1s 45 derece 5 <1s uyarıya normal 0 >1s uyarıya hipoaktif 1 BİLİNÇ ROTASYON TESTİ AĞRI YANITI TOPLAM 0-20 arası Tablo 6: Modifiye Bederson nörolojik muayene skorlaması (9, 100) 38 4.4. Ölçülen Parametreler 4.4.1. Beyin Ödemi Beyin ödeminin değerlendirilmesi için beynin ıslak/ kuru su içeriği oranından yararlanılmıştır (100). Bu amaçla dekapitasyon ile çıkarılan beyinler darası alınan bir porselen kapsüle konularak hassas terazide (Mettler AE50) tartılır. Daha sonra 1000C etüvde 24 saat kurutulup tekrar tartılır. % su içeriği= (ıslak-kuru)/ ıslak x100 formülü ile hesaplanır. 4.4.2. KBB Geçirgenliği Travma oluşturulduktan sonraki 48. saatte ketamin-klorpromazin anestezisi altında sıçanlara juguler venden %2’lik evans mavisi (EB) çözeltisi 4ml/kg dozda verilerek 30 dakika beklenmiştir (Bkz. Resim 4). Daha sonra damar içindeki boya transkardiyak (sol ventriküle girilip, sağ atriyuma çentik atılır) olarak verilen 110mmHg basınçlı yaklaşık 250 ml SF perfüzyonu ile 15 dakika yıkanmıştır. Sağ atriyumdan berrak sıvı geldiğinde perfüzyon sonlandırılmıştır. Beyinler hemen çıkarılarak tartıldı. Örnekler 2.5 ml pH 7.4 fosfat tamponu ile homojenize edilerek aşağıdaki prosedüre göre çalışıldı (Toklu ve ark. 2008). Gerekli Çözeltiler: Standart Çözeltisi: Evans mavisinin (Sigma E2129) 10 µg/ml’lik stok çözeltisinden farklı seyreltmeler yapılarak standart çözelti hazırlandı. 0.1M Fosfat tamponu: 13.6g KH2PO4, 14.4g Na2HPO4, 2g KCl ve 9g NaCl suda çözülerek 1000 ml’ye tamamlandı. Daha sonra pH 7.4’e ayarlandı. % 60 TCA çözeltisi Deneyin Yapılışı: Kör Standart 2.5 ml pH 7.4 fosfat tamponu 2.5 ml standart çözeltisi Numune 2.5ml numune + 2.5 ml %60 TCA Vorteks (Ika-VF2) ile karıştırıldı. 39 Üst fazda çalışıldı, çökelti atıldı. Buzdolabında 40C’de 30 dakika bekletildi. 30 dakika 1000 G devirde santrifüj (Hettich Universal) edildi. Buzdolabında 40C’de 30 dakika bekletildi. 30 dakika 1000 G devirde santrifüj (Hettich Universal) edildi. Üst faz alınarak oluşan rengin absorbansı spektrofotometrede (Shimadzu UV-1208) 620 nm’de okundu. 40 Resim 4: Evans mavisi enjeksiyonu takiben 30 dakikalık bekleme sürecindeki sıçanlar 41 4.4.3. Oksidan Hasar ve Antioksidan Savunma Travma oluşturulduktan 48 saat sonra dekapite edilen sıçanların beyin dokusunda oksidan hasarın bir göstergesi olarak lipid peroksidasyonu tiyobarbitürik asitle reaksiyona giren madde MDA miktarı üzerinden ölçülmüştür. Antioksidan savunma mekanizması ise indirgenmiş glutatyon GSH üzerinden ölçülmüştür. 4.4.4. MDA Lipid peroksidasyonunun göstergesi olan beyin dokularındaki MDA miktarı, Buege yöntemine göre tayin edilmiştir (11). Dekapitasyondan hemen sonra çıkarılan beyinler serum fizyolojik ile yıkandıktan sonra filtre kağıdı ile kurutuldu, tartıldı. Ika Werk homojenizatöründe buz üstünde 150 mM KCl çözeltisiyle %10’luk homojenatı hazırlandı. Gerekli Çözeltiler TBA (2-Tiyobarbitürik asit, Sigma T5500 ) çözeltisi: % 0.375 g TBA ve % 15’lik TCA (Trikloroasetik asit) içeren çözelti, 0.25N HCl (Hidroklorik asit) ile hazırlandı. Standart Çözeltisi: 1,1,3,3 tetraetoksipropan (malonaldehid bis [dietil asetal], Sigma T9889)’ın 10 mM’lık stok çözeltisinden farklı seyreltmeler yapılarak standart çözelti hazırlandı. Deneyin Yapılışı: Kör 0.5 ml 150 mM KCl Standart 0.5 ml standart çözeltisi Numune 0.5 ml böbrek homojenat 1 ml % 0.375 TBA 15 dakika kaynar su banyosuna bırakıldı ve sonra tüpler oda sıcaklığında soğutuldu 42 10 dakika 3000 devir/dakikada santrifüj (Hettich Universal) edildi. Üst faz alınarak oluşan pembe rengin absorbansı spektrofotometrede (Shimadzu UV–1208) 532 nm’de okundu. 4.4.5. GSH Dokuda GSH tayini Ellman yöntemine göre yapıldı (12, 13). GSH tayininde MDA tayini için hazırlanan % 10’luk homojenatlar kullanıldı. Gerekli Çözeltiler: 0,3M Na2HPO4 (Disodyum hidrojen fosfat, Carlo Erba 480137) 10mM DTNB (Ditiyo nitro benzen, Ellman, Sigma D8130): 39.63 mg DTNB % 1’lik sodyum sitrat ile 10 ml’ye tamamlandı. % 20’lik TCA çözeltisi : Trikloro asetik asit (Carlo Erba 411525) Standart glutatyon çözeltisi: 50 mg glutatyon (Sigma G4251) distile suda çözüldü, 5 ml’ye tamamlandı. Bu stoktan farklı seyreltmeler yapılarak standart çözelti hazırlandı. Deneyin Yapılışı: 0.4 ml % 10’luk homojenat + 0.2 ml % 20’lik TCA Karıştırıldı. 15 dakika 3000 devir/dakikada santrifüj (Hettich Universal) edildi. Üst fazda GSH çalışıldı, çökelti atıldı. 43 Kör Standart 0.2 ml 150mM KCl 0.2 ml standart çözeltisi Numune 0.2 ml üstfaz 1 ml 0.3 M Na2HPO4 + 0.05 ml Ellman çözeltisi ilave edilerek karıştırıldı. 5 dakika bekleildi ve oluşan sarı rengin absorbansı spektrofotometrede (Shimadzu UV–1208) 412 nm.’de okundu. 4.4.6. MPO Dokuda Myeloperoksidaz (MPO) Aktivitesi Tayini: Dokuda MPO tayini Hillegas yöntemi ile yapıldı (47). Dekapitasyondan hemen sonra çıkarılan serebral doku, kan ve çevre artıklarından temizlenmesi için serum fizyolojik ile yıkandıktan sonra filtre kağıdı ile kurutuldu ve ağırlıkları alındı. Böbrek dokusu 50 mM K2HPO4 (pH : 6 ) ile homojenize edildi. % 10luk homojenat hazırlandı. Gerekli çözeltiler 50 mM K2HPO4 (pH : 6 ) % 0.5’lik HETAB (Hekzadesiltrimetil-amonyum bromid) : 0.5 g HETAB, 50 mM K2HPO4 ile çözüldü. pH 6’ya ayarlandı ve hacmi 50 mM K2HPO4 ile 1 lt’ye tamamlandı. o-Dianizidin-2 HCl: 100 mg o-Dianizidin-2HCl distile suda çözülerek hacmi 5 ml’ye distile su ile tamamlandı. %2’lik sodyum azid 20 mM H2O2 (Hidrojen peroksit) Deneyin Yapılışı : 3 ml % 10 luk böbrek homojenatı 41400 g de 4 °C lik sıcaklıkta 10 dakika santrifüj edildi. 44 Üst faz atıldı, çökelti + 3 ml % 0.5’lik HETAB Homojenizasyon 3 kez donduruldu – çözündürüldü – sonike edildi 41400 g de 4 °C lik sıcaklıkta 10 dakika santrifüj edildi. 0.3 ml üst faz ile çalışıldı. Bu aşamadan sonraki işlemler 37 °C lik su banyosunda yapıldı. Kör Numune 2.9 ml 50 mM K2HPO4 (pH: 6) 2.9 ml 50 mM K2HPO4 (pH: 6) 0.2 ml 20 mM H2O2 0.2 ml 20 mM H2O2 0.2 ml o-Dianizidin-2 HCl 0.2 ml o-Dianizidin-2 HCl 0.3 ml 50 mM K2HPO4 (pH: 6) 0.3 ml üst faz 3 dakika beklendi. . 0.2 ml % 2’lik sodyum azid ile renk reaksiyonu durduruldu 37°C lik su banyosundan alındı, karıştırıldı. Oda sıcaklığında bekletildi. 3000 devir/dakikada 10 dakika santrifüj edildi. Üst faz alınarak oluşan rengin absorbansı spektrofotometrede 460 nm de okundu. 45 4.4.7. Na+ K+-ATPaz Tayini Reading yöntemine göre yapılmıştır (81). 0.32 M sukroz çözeltisi ile % 10’luk homjenat hazırlanır. 3000 rpm’de 5 dakika santrifüj edilir. KÖR A çözeltisi Mg+2-ATP az TOTAL ATPaz 0.75 ml 0.75 ml - B çözeltisi - - 0.75 ml Ouabain - - 0.5 ml Distile su 0.5 ml 0.5 ml - 0.1 ml 0.1 ml - Üst faz Distile su 0.1 ml - 37 ° C’de 5 dk inkübasyon ATP 0.1 ml 0.1 ml 0.1 ml 0.1 ml 0.1 ml 37 ° C’de 10 dk inkübasyon TCA 0.1 ml A çözeltisi 0.58 g NaCl, 0.037 g KCl, 0.122 g MgCl2, 0.0037 g EDTA, 0.472 g Tris-HCl 100ml distile suda çözülerek pH 7.4’e ayarlanır. B çözeltisi 0.061g MgCl2 50 ml distile suda çözülür. ATP çözeltisi 0.206 g disodyum ATP distile suda çözülerek 5ml’ye tamamlanır. 46 FOSFOR TAYİNİ Fiske ve Subbarrow yöntemine göre yapılmıştır (34). Std Körü Std Num, körü Total ATP az Mg-ATPaz 0.5 ml TCA 0.5 ml std. 0.5 ml üst faz 0.5 ml üst faz 0.5ml üst faz 1 ml amonyum molibdat çözeltisi 0.4 ml Fiske-subbarow çözeltisi 3.1 ml distile su 5 dk. beklet Spektrofotometrede 690 nm’de oku. PROTEİN TAYİNİ Protein tayini Lowry metodu (63) ile yapılmıştır. Kör 0.5 ml serum fizyolojik Standart Numune 0.5 ml standart protein çözeltisi 0.01ml böbrek homojenatı + 0.49 ml serum fizyolojik + 3 ml C çözeltisi karıştırıldı ve 10 dakika oda sıcaklığında bekletildi. + 0.1 ml folin çözeltisi karıştırıldı ve 30 dakika bekletildi. 47 Oluşan rengin absorbansı spektrofotometrede 500 nm de okundu. C protein çözeltisi için 50ml Protein çözeltisi I, 1ml Protein Çözeltisi II ile karıştırılır. Protein Çözeltisi I 2g NaHCO3 0.1N NaOH içinde çözülerek 100ml’ye tamamlanır. Protein Çözeltisi II % 1 CuSO4 çözeltisi, % 2 Na-K-tartarat çözeltisi ile 1:1 oranında karıştırılır. 4.4.8. Histolojik incelemeler ve perfüzyon fiksasyonu Perfüzyon aparatı bir litrelik iki adet intravenöz serum şişesi ve bunlara bağlı üç yollu iv kanülden oluşmaktadır. (Caire klempli Venoset; 150 cm., 15 damla/ml). İntravenöz serum şişeleri sıçanların perfüze edileceği tezgahın yaklaşık 120-125 cm yukarısına asıldı. Şişelerden birisinde %0.9 sodyum klorür diğerinde ise PBS tamponu içinde %2,5’lik glutaraldehit kondu (pH = 7.4)). Üçlü vananın çıkışına, distal ucunda 18-gauge iğne bulunan, iç çapı 3.0 mm ve boyu 15-30 cm olan propilen bir boru bağlandı (Şekil 4). Şekil 4: Perfüzyon düzeneği. Iki adet birer litreli serum şişesinden (A), birinde serum fizyolojik, diğerinde ise tamponlu glutaraldehit bulunmaktaydı. Bu şişeler üç yollu vanaya (C) takılmış bir iv sete (B) bağlanmıştı. Şişeler tezgahtan 120 cm yukarıya asıldı. Vana çıkışına bağlı tüpün uzunlığı 15-30 cm arasında tutuldu (D). Bu tüpün ucuna da 18-guage iğne (E) bağlandı. 48 Sıçanların Hazırlanması Perfüzyon tespitinini başlamasından 15 dakika önce, 300 gramdan daha ağır olan sıçanlara anestezi öncesi 150U/kg intraperitoneal heparin verilerek 10 dk süreyle etkisinin oluşması beklendi. Daha sonra intraperitoneal uygulanan 2 mg/kg ketamin ve klorpromazin ile anestezi gerçekleştirildi. Anestezinin gerçekleştiği doğrulandıktan sonra sıçanların ekstremiteleri ve kuyrukları kesim panelleri üzerine geniş yapışkan bantlarla tespit edildi. V kesi ile sternum kaldırılarak toraks boşluğu açıldı. Kalbin tepesinden 18 gauge kelebek iğne ile girilerek sol ventriküle dikkatli bir biçimde yerleştirildi. Daha sonra sağ atriyum kesildi ve üç yollu kanülden önce 10 dk süreyle % 0,9 NaCl verildi. Sağ atriyumdan akan sıvının berraklaştığı görüldüğünde kanülün vanası diğer girişe çevrilerek fosfat tamponu içinde % 2.5 glutaraldehid çözeltisi 35 damla/15 sn olacak şekilde verilmek suretiyle 25 dk süreyle perfüzyon tespiti gerçekleştirildi. Her sıçan için yaklaşık 300 ml tespit sıvısı kullanıldı 8, 72). 4.5. İstatistiksel Değerlendirme Bu çalışmanın verileri ortalama ± standart hata olarak verilmiştir. Tüm testlerde P<0.05 hata istatistiksel olarak anlamlı kabul edilmiştir. Testler bilgisayarda Graph-Pad Prism 4.0 (GraphPad Software, 2003, San Diego; CA; USA) bilgisayar istatistik programı kullanılarak yapılmıştır. Grupların karşılaştırılmasında ise tek yönlü ANOVA testi kullanılmıştır. Doğrulama testi olarak da Tukey’in çoklu karşılaştırma testi yapılmıştır. Nörolojik muayene skorlaması için nonparametrik Kruskal-Wallis Testi ve takiben Dunn’s çoklu karşılaştırma testi kullanılmıştır. 49 5. Bulgular 5.1 Nörolojik Muayene Dekapitalize edilmeden önce yapılan nörolojik muayene skorlamasında travmaya göre montelukast verilen sıçanların nörolojik muayene skorlarının daha iyi olduğu gözlenmekle 10 *** 8 * 6 4 2 tr au m a+ m on te tr au m a nt co m on te 0 ro l neurological examination score birlikte istatistiksel olarak anlamlı bir fark yoktur (Şekil 5). Şekil 5: Bederson nörolojik muayene skorlamaları. Gruplar 6-8 hayvandan oluşmaktadır. Kruskal-Wallis çoklu karşılaştırma testini takiben doğrulama testi olarak Dunn’s testi kullanılmıştır. *p<0.05, *** p<0.001 kontrole göre. 50 5.2. MDA ve GSH Lipid peroksidasyonunun ve oksidan hasarın bir göstergesi olan MDA düzeyi travma oluşturulmasını takiben artış gösterirken (p<0.001), GSH düzeyi de paralel olarak azalmıştır (p<0.001). Montelukast uygulaması lipid peroksidasyonunu anlamlı olarak azaltmıştır. Ancak GSH düzeylerine etki etmemiştir. (Şekil 6a ve 6b) a) 60 *** MDA (nmol/g) 50 + 40 30 20 10 m on te a um tr a um a+ tr a m on te co nt ro l 0 b) GSH (μmol/g) 2.0 1.5 *** 1.0 0.5 on te a tr au m a+ m m tr au on te m co nt ro l 0.0 Şekil 6: Deney hayvanlarının travma sonrasında beyin dokusunda a) malondialdehid (MDA), b) Glutatyon (GSH) içeriği. Gruplar 6-8 hayvandan oluşmaktadır. Tek yönlü ANOVA testi; ***: p<0.001, kontrol grubu ile karşılaştırma; +: p<0.05, travma ve travma+montelukast grubunun karşılaştırılması 51 5.3. MPO ve Na-K-ATPaz Travma sonrası myeloperoksidaz aktivitesinde anlamlı bir artış görülürken (p< 0.005) Na+ K+-ATP az etkinliğinde anlamlı azalma tespit edilmiştir (p< 0.001). Montelukast uygulamasını takiben myeloperoksidaz aktivitesinde anlamlı azalma olmuştur (Şekil 7a). Ancak Na+ K+ az etkinliğinde fark bulunamamıştır. (Şekil 7b) a) MPO activity (U/g) 9 * + 6 3 tr au m a+ m on te a tr au m m on te co nt ro l 0 b) Na-K-ATPase activity ( μmol/mg/h) 10.0 7.5 5.0 *** 2.5 on te tr au m a+ m a tr au m on te m co nt ro l 0.0 Şekil 7: Deney hayvanlarının travma sonrasında beyin dokusunda a) Myeloperoksidaz (MPO), b) NA+ K+ az tayini. Gruplar 6-8 hayvandan oluşmaktadır. Tek yönlü ANOVA testi; * p<0.05, ***: p<0.001, kontrol grubu ile karşılaştırma; +: p<0.05, travma ve travma+montelukast grubunun karşılaştırılması 52 5.4. Kan beyin bariyeri geçirgenliği Evans mavisi ekstravazasyonu ile değerlendirilen kan beyin bariyeri geçirgenliğinde travma sonrası makroskopik olarak da gözlenebilen bir artış ortaya çıkmıştır. (Resim 5, 6) Makroskopik olarak gözlenen bu boyanma kesitsel olarak incelendiğinde daha da belirgindir. Geçirgenlikteki değişmenin kantitatif tayininde de travma grubunda anlamlı (p<0.001) bir artış saptanmıştır. Montelukast tedavisi sonrası evans mavisi ekstravazasyonu halen anlamlı (p< 0.05) olarak yüksek olmakla birlikte travma grubu ile karşılaştırıldığında anlamlı (p< 0.05) olarak azalmıştır (Şekil 8a). Ödem açısından ise tedavi ve travma grupları arasında bir fark bulunmamıştır (Şekil 8b). SF montelukast kontrol travma Resim 5: Evans mavisi enjeksiyonu sonrası makroskopik görünüm SF montelukast kontrol travma Resim 6: Kesitsel görünüm 53 a) 5 *** EB mg/g tissue 4 * + 3 2 1 on te a tr au m a+ m tr au m m on te co nt ro l 0 b) brain water content % 100 95 90 * 85 80 75 te tr au m a+ m on a m tr au m on te co nt ro l 70 Şekil 8: Deney hayvanlarının travma sonrasında beyin dokusunda a) Evans mavisi ekstravazasyonu, (b) Beyin su içeriği. Gruplar 6-8 hayvandan oluşmaktadır. Tek yönlü ANOVA testi; * p<0.05, ***: p<0.001, kontrol grubu ile karşılaştırma; +: p<0.05, travma ve travma+montelukast grubunun karşılaştırılması 54 5.5. Histolojik İncelemeler kontrol a a travma b monte c Resim 6: A) Elektron mikroskopik değerlendirme ile kontrol grubunda nöronların ince yapısı ve dendritik, aksonal, glial çıkıntıların karmaşık görünümünün sağlam olduğu görüntülenmiştir. Aksonal çıkıntıların etrafındaki miyelin kılıf, beyin bariyeri ve nörofiller sağlam görünmektedir. B) Travma grubunda nöron hücre gövdeleri ve nörofillerde (aksonlar, dendritler ve glial çıkıntılar) hidrofik dejenerasyonun bir bulgusu olarak şiddetli şişme gözlemlenmiştir. C) Astrositler ve endotel hücrelerinde ödem olsa da nukleolus sağlamdı ve nukleusta kromatin dağılması gözlemlenmedi. 55 6. Tartışma ve Sonuç Gelişmiş modern tanı ve tedavi yöntemlerine karşın, kafa travmalarına bağlı ölüm günümüzde hala problem oluşturmaktadır (23). Kafa travmalarının en sık nedeni trafik kazalarıdır. Düşmeler, spor yaralanmaları, iş kazaları, darp ve terör yaralanmaları diğer nedenlerdir. Travmatik beyin yaralanması primer ve sekonder beyin yaralanlaması olarak ikiye ayrılır (41). İnsanda ki kafa travmasının primer ve sekonder sebeplerini araştırmak ve insan beyni mevcut sinir hasarını onarmaya yönelik çeşitli tedavi yöntemlerini geliştirmek için kafa travması hayvan modelleri yapılmaktadır. Deneysel modellerin temel amacı nekrotik ve apoptotik nöronal hücre ölümünü ortaya koymaktır (46). Temel olarak iyonik dengelerin bozulması, ortaya çıkan serbest radikaller, enflamatuar ve immunolojik cevap, ekstitatör aminoasitlerin salınması, nörotransmitter ve nöromodülatör sistemlerin bozulması olarak sayılabilir (19). Deneysel kafa travmaları, Denny-Brown Russell’in çalışmalarıyla akselerasyon konküzyon ve perküsyon-konküzyon olmak üzere iki kategoriye ayırmıştır ( 28). Çalışma için seçilen deneysel model, diğer modellere göre uygulanması kolay, ucuz, kontrol edilebilir bir model olan Marmarou’nun impakt akselerasyon modelidir. Bu model hem apopitozik hem de nekrotik hücre ölüm hasarı ortaya çıkarmaktadır. Histopatolojik olarak yaygın, bilateral nöron, akson, dentrit ve mikrovasküler hasarı ve diffüz aksonal yaralanma oluşur. İlk 4 saatde serebral kan akımında azalma ve intrakranyal basınçda artma ile sekonder otoregülasyon bozukluğu oluşur. İlk saatlerde vazojenik ödem daha sonra yaygın hücresel şişme meydana gelişir. İlk 20 dakikasında başlayan ve 24 saatten sonra devam eden beyin ödemi oluştuğu, ilk 4 saatten sonra kan beyin bariyeri geçirgenliğini bozulduğu gösterilmiştir (19, 68). Travmatik beyin yaralanamasının farmakolojik tedavisi antienflamatuvarlar, glutamat antagonistleri, katyon homeostazı modülatörleri, endokannobinoidler, serbest radikal temizleyicileri, immünsupresanlar, apopitoz ve kaspaz inhibitörlerleri, nörotropik faktörler kullanılmaktadır (86). Travmadan sonra beyin kan akımında azalma ile doku oksijen düzeylerinde iskemiye neden olacak kadar düşüş ve sonuçta sekonder yaralanma ve beyin ödemine yol açan ilerleyici fizyopatolojik değişikler olduğuna ilişkin bulgular vardır. Çok sayıda klinik ve deneysel çalışma sekonder beyin yaralanmasının TBY’na yanıt olarak sıklıkla artan serbest radikal oluşumu, enflamasyon, adhezyon molekülleri, sitokin ve kemokinlerin ortaya çıkması, ile 56 sekonder beyin yaralanmasının daha da ilerlediğini göstermiştir (56, 74). Yaralanmış beyin dokusunda IL-1β, TNF-α, IL-6 ve intersellüler adezyon molekülü-1 gibi moleküllerin serebral hasar, hücre ölümü ve KBB disfonksiyonuna katkıda bulunduğuna inanılmaktadır (2). Travmaya sekonder olarak serebral iskemi ve beyin ödemi gelişir. Arişidonik asidin 5lipooksijenaz metoboliti olan sisteinil lökotrienler travma yanısıra iskemi, tümör, MS, ensefalomyelit ve yaşlanma gibi çeşitli SSS hastalıklarında da artar (94). Artan CysLT’ler kan-beyin bariyer disfonksiyonu ve beyin ödemi ile uyumludur. Bu nedenle CysLT’lerin beyinde birikiminin serebral iskemi anahtar rol oynayabilecaği düşünülmektedir (107). Akut enflamasyonun özelliği olarak dokuya olan PMN lokösit infiltrasyonu kemotaktik mediatörlerin kolektif etkisinin var olduğunu işaret etmektedir. Nötrofiller iskemik alana göç ettiğinde reaktif oksijen ürünleri, proteazlar, elastazlar, myeloperoksidaz, sitokinler ve doku zedelenmesinde yer alan çeşitli diğer sitokinleri salarlar (87, 88, 110). Çeşitli kemokinler ve bir lipit mediatör metoboliti olan Sisteinil LT’ler iskemi oluşmasına sebep olan güçlü inflamatör mediatörlerdir (107). Yapılan araştırmalarda sıçan modelinde SisteinilLT lerin beyin travması sonrasında yükseldiği, 4. saat ve 7. günde pik yaptığı, bunun da ödeme ve hücresel enflamatuvar yanıta bağlı olduğu bildirilmiştir (108). Bu çalışma sıçanlarda travmatik beyin hasarında gelişen sekonder yaralanmada Sisteinil LT reseptör antagonisti montelukastın etkisini araştırmak için yapılmıştır. Montelukast eozinolotik enflamasyonu bu hücrelerden enflamatuvar sitokin salınımını inhibe eder (73). Orta şiddetli beyin yaralanmasının yaygın lipit peroksidasyonunu indüktediğini göstermişlerdir. (80) Oluşturulan deneysel kafa travması, 48. saatte dekapitalize edilen hayvanlarda lipid peroksidasyonun ve oksidan hasarın göstergesi olan MDA nın anlamlı olarak artmıştır. Bu artışa GSH düzeylerinde paralel olarak azaldığı ve dokuya nötrofil infiltrasyonu olduğunu işaret eden MPO artış eşlik etmiştir. Montelukast tedavisi sisteinil LT1 antagonizması üzerinden antienflamatuar etkileri ile MDA ve MPO’yu belirgin olarak hemen hemen kontrol düzeylerinin altına çekmiştir. Bu nedenle, montelukastın etkisinin beynin endojen antioksidan içeriğini etkilemediği için antioksidan etkinliğe değil antienflamatuar etkinliğe bağlı görünmektedir. Montelukastın GSH düzeylerini korumak, MPO ve MDA’yı düşürmek sureti ile antioksidan dengeyi sürdürerek iskemi reperfüzyon yaralanmasına karşı faydalı etki ortaya koyduğu daha önce gösterilmiştir (90, 91). GSH düzeyleri travma grubunda daha yüksek görünse de istatistiksel olarak anlamlı değildir. Ancak bu çalışmada yalnızca antienflamatuvar 57 etkisiyle oksidan hasarı azalttığını, antioksidan savunma sistemine etki etmediğini söyleyebiliriz. Ya da hasar o kadar fazla oluşmuş olabilir ki, GSH çok fazla tükenmiş ve montelukast buna yetişememiş olabiliir. Travmadan sonra ortaya çıkan ilerleyici fizyopatolojik değişikliklerin beyin kan akımında azalmaya, iskemiye yol açan doku oksijen seviyelerinde düşmeye ve sonuçta da sekonder yaralanmaya ve beyin ödemine neden olduğuna ilişkin bulgular vardır. İskemik durumlar ve TBY’na bağlı SSS patolojilerinde Na-K ATPaz yetmezliği sık görülür (105, 62). Düşük O2 ‘de Na/K ATPaz net şekilde azalır ve bu durum beyin ödemini tetikleyerek KBB’nin yıkılmasına yol açar (4, 78, 102) TBY’nin pompa aktivitesinde anlamlı inhibisyon yaptığını ve oksidatif stres ile yakından ilişkili olduğunu gösteren sonuçlar mevcuttur. Montelukast, pompa fonksiyonunun yeniden kazanılmasında etkisiz olduğu bulunmuştur. Bu nedenle Montelukast’ın ödeme de etkisiz olduğu, dolayısıyla ödemi azaltmamasına Na/K ATPaz pompasına etkili olmamasından kaynaklanacağı ileri sürülebilir. Önceki çalışmalar ile tutarlı olarak TBY pompa aktivitesinde montelukast tedavisi ile düzelmeyen anlamlı inhibisyona neden olmuştur. Nörolojik muayene skorları da montelukast tedavisi ile düzelmemiştir. Lima ve çalışma arkadaşlarının belirttiği gibi montelukast tedavisi ile nörolojik durumun korunmaması, pompa fonksiyonunu düzeltememesine ve beyin ödemi oluşumunu önleyememesine bağlı olabilir. Montelukast tedavisi beyin ödemi oluşumunun azaltılmasında etkisiz olmuşsa da KBB geçirgenliğini anlamlı olarak azaltabilmiştir (62). EB’nin ekstravazasyonu kontrollerden anlamlı derecede fazla olmuştur. Enflamatuvar mediyatörlerin KBB’nin yıkılmasını arttırdığı iyi bilinmektedir. Montelukast, muhtemelen hem serbest radikalleri hem de bunların antienflamatuvar etkilerini azaltarak KBB geçirgenliğini azaltmaktadır. Montelukast tedavisi KBB geçirgenliğini tam değil kısmen azaltıyor görünmekte, histolojik değerlendirme ile de gösterildiği gibi KBB bütünlüğünü tam sağlayamamaktadır. Histolojik bulgular travmanın hem vazojenik hem de sitotoksik beyin ödemine neden olduğunu doğrulamaktadır. Ayrıca travma hücresel elemanlarda sitotoksik ödemin varlığına işaret eden şişmeye de sebep olmuştur. Vazojenik ödemde KBB’nin yıkılması plazma benzeri sıvının ekstrasellüler boşluğa ekstravazasyonuna izin vermektedir. Diğer taraftan, sitotoksik ödem esas olarak Na/K-ATPaz akativitesinin bozulmasına bağlı ekstrasellüler boşluğun daralması ile karakterizedir (4). Bozulan pompa fonksiyonunu düzeltemediği için monteklukastın sitotoksik ödeme karşı etkisiz olduğu ileri sürülebilir. Histolojik bulgularımızın biyokimyasal bulgularımız ile tutarlı görünmektedir. 58 Yapılan çalışmalarda nötrofil perfüzyonuna uğramış beynin formillenmiş tripeptide maruz bırakılmasının anlamlı sisteinil LT artışının sonucu olduğu ve kan-beyin bariyerinde bozulma ile sonuçlandığını bulunmuştur. Çift yönlü sisteinil LT1 ve sisteinil LT2 reseptör antagonisti ile tedavinin beyin geçirgenliğini önlemede tek yönlü sisteinil LT1 reseptör antagonisitine göre daha etkili olduğunu gösterilmiş ve elde edilen snuçların beyin damar yapısında endotel ve nötrofillerin sisteinil LT’lerin lokal sentezinde birlikte yer aldığı ve bunun da serebral ödem oluşumundaki patojenik mekanizmalara katkıda bulunduğunu öne sürülmüştür (30) Beyin ödemi oluşumunu arttıran enflamatuvar mediyatörler, serbest radikaller, proteazlar, VEGF, bradikinin, araşidonat metabolitleri gibi çok sayıda faktör vardır (4, 102). Başlangıçtaki yüksek LT düzeyinin vazojenik ödem bileşenin arttırmaktan çok sitotoksik ödeme katkıda bulunduğu daha önce gösterilmiştir (89). CysLT1 reseptörlerinin mikrovasküler endotel hücrelerinde bulunduğu daha önce gösterilmiştir. Beyindeki hakim CysLT2 reseptörü TBY’nda ve beyin tümöründe nöronlarda ve glia hücrelerinde indüktlenmektedir (108). Montelukastın ödemi etkileyememiş olmasının nedeni seçici sisteinil LT1 antagonizması özelliğine bağlı olabilir. Yakın zamanda LTD4’ün sitotoksik ödemin AQP4 (akuaporin 4) aracılı CysLT2 reseptörü yolu ile indüklediği gösterilmiştir (104). Sheng ve ark (93) CysLT1 ve CysLT2 reseptörlerinin oksijen-glukoz tükenmesinin indüklediği hücre ölümünde de farklı rolleri olduğunu göstermişlerdir. Vurgulanan çalışmada sisteinil LT1 antagonisti montelukast apopitozu etkilemezken seçici olmayan sisteinil LT antagonisti olan Bay u9773 ise özellikle CysLT2 reseptörlerinin fazla olduğu hücrelerde apopitozu inhibe etmiştir (93). CysLT1 ve CysLT2 üreten 5-lipooksijenazın astrositlerde iskemi tarafından aktiflenmesinin CysLT1 aracılıklı proliferasyona ve CysLT2 reseptör aracılıklı ölüme yol açtığını göstermiştir (49). İki reseptörün farklı rollerinin olması monteukastın bu çalışmada değerlendirilen bazı parametreler üzerinde farklı etkiler göstermesini açıklayabilir. Daha önce iddia ettiğimiz üzere histolojik bulgularımızın biyokimyasal bulgularımız ile tutarlı olduğu düşünülebilir. Histolojik gözlemlere göre travma, KBB’nin yıkılmasının yanısıra ödem ve nöron dejenerasyonuna da neden olmaktadır. Montelukast ile tedavi edilen sıçanların beyinlerinde nukleolusun sağlam olduğu ve kromatin dağılmasının olmadığı görülmüştür. Diğer araştırmacılar da montelukastın fokal serebral iskemi modeli (107) ve spinal kord yaralanması modelinde (38) nöroproteksiyon etki yaptığını göstermişlerdir. 59 Sonuç olarak TBY’dan sonraki 48 saat içinde uygulanan montelukast tedavisinin nöron dejenerasyonunu önlediği ve yavaşlattığı ileri sürülebilir. Bu etki, muhtemelen montelukastın antienflamatuvar özelliklere bağlıdır. Bozulan Na/K-ATPaz aktivitesinin korunmasında ve sitotoksik ödem oluşumunun önlenmesinde etkisiz olmuştur. Na/K-ATPaz pompasının yetersizliği ile yakından ilişkili olduğu gösterilen nörolojik muayene skorları bu sebeple düzelmemiştir. Bu çalışmanın hassas noktası, etkilerinin tek bir zaman noktasında değerlendirilmiş olmasıdır. Montelukastın zamana bağlı etkilerini değerlendirmek ve TBY’ndaki etki mekanizmasını aydınlatılabilmek için başka çalışmalar yapılmalıdır. Montelukastın etkilerinin diğer sisteinil LT2 antagonistleri ile karşılaştırılması da faydalı olabilir. 60 7. Kaynaklar 1. Allan SM, Rothwell NJ. Inflammation in central nervous system injury. Philos Trans R Soc Lond B Biol Sci 358(1438): 1669–1677, 2003. 2. Ansari MA, Roberts KN, Scheff SW. Oxidative stress and modification of synaptic proteins in hippocampus after traumatic brain injury. Free Radic Biol Med 45(4): 443– 452, 2008. 3. Armstead WM. Differential activation of ERK, p38, and JNK MAPK by nociceptin/orphanin FQ in the potentiation of prostaglandin cerebrovasoconstriction after brain injury. Eur J Pharmacol 529: 129–135, 2006 4. Ayata C, Ropper AH. Ischaemic brain oedema. J Clin Neurosci 9(2): 113–124. 2002 5. Bäck M. Leukotriene Receptors: Crucial Components in Vascular inflammation. ScientificWorldJournal 7: 1422–1439, 2007 6. Balani SK, Xu X, Pratha V, Koss MA, Amin RD, Dufresne C, Miller RR, Arison BH, Doss GA, Chiba M, Freeman A, Holland SD, Schwartz JI, Lasseter KC, Gertz BJ, Isenberg JI, Rogers JD, Lin JH, Baillie TA. Metabolic profiles of montelukast sodium (Singulair), a potent cysteinyl leukotriene1 receptor antagonist, in human plasma and bile. Drug Metab Dispos 25(11): 1282–1287, 1997 7. Bayir H, Kagan VE, Borisenko GG, Tyurina YY, Janesko KL, Vagni VA, Billiar TR, Williams DL, Kochanek PM. Enhanced oxidative stress in iNOS-deficient mice after traumatic brain injury: support for a neuroprotective role of iNOS. J Cereb Blood Flow Metab 25: 673–684, 2005 8. Beach TG, Tago H, Nagai T, Kimura H, McGeer PL, McGeer EG. Perfusion-fixation of the human brain for immunohistochemistry: comparison with immersion-fixation. J Neurosci Methods 19(3):183-92, 1987. 9. Bederson JB, Pitts LH, Tsuji M, Nishimura MC, Davis RL, Bartkowski H. Rat middle cerebral artery occlusion: evaluation of the model and development of a neurologic examination. Stroke 17(3): 472–476, 1986 10. Bernstein PR. Chemistry and structure-activity relationships of leukotriene receptor antagonists. Am J Respir Crit Care Med 157: 220–225, 1998 11. Beuge JA, Aust SD. Microsomal lipid peroxidation. Methods Enzymol 52: 302–311, 1978 12. Beutler E, Duron O, Kelly BM. Improved method for the determination of blood glutathione. J Lab Clin Med 61: 882–888, 1963 61 13. Beutler, E. Glutathione in red blood cell metabolism. A Manuel of Biochemical Methods, Grune & Stratton New York s.112-114, 1975 14. Born JD, Albert A, Hans P, Bonnal J. Relative prognostic value of best motor response and brain stem reflexes in patients with severe head injury. Neurosurgery 16(5): 595–601, 1985 15. Bouma GJ, Muizelaar JP. Cerebral blood flow, cerebral blood volume, and cerebrovascular reactivity after severe head injury. J Neurotrauma 9: 333–348, 1992 16. Bramlett HM, Dietrich WD. Pathophysiology of cerebral ischemia and brain trauma: similarities and differences. J Cereb Blood Flow Metab 24: 133–150, 2004 17. Bullock R, Zauner A, Woodward JJ, Myseros J, Choi SC, Ward JD, Marmarou A, Young HF. Factors affecting excitatory amino acid release following severe human head injury. J Neurosurg 89: 507–518, 1998 18. Capra V, Ambrosio M, Riccioni G, Rovati GE. Cysteinyl-leukotriene receptor antagonists: present situation and future opportunities. Curr Med Chem13(26): 3213– 3226, 2006 19. Cernak I. Animal models of head trauma. NeuroRx 2(3): 410–422, 2005 20. Chong ZZ, Li F, Maiese K. Oxidative stress in the brain: Novel cellular targets that governsurvival during neurodegenerative disease. Prog Neurobiology 75: 207–246, 2005 21. Ciana P, Fumagalli M, Trincavelli M.L, Verderio C, Rosa P, Lecca D, Ferrario S, Parravicini C, Capra V, Gelosa P, Guerrini U, Belcredito S, Cimino M, Sironi L, Tremoli E, Rovati G.E, Martini C, and Abbracchio MP. The orphan receptor GPR17 identified as a new dual uracil nucleotides/cysteinyl-leukotrienes receptor. EMBO J 25: 4615–4627, 2006 22. Coles JP, Fryer TD, Smielewski P, Rice K, Clark JC, Pickard JD, Menon DK. Defining ischemic burden after traumatic brain injury using 15O PET imaging of cerebral physiology. J Cereb Blood Flow Metab 24: 191–201, 2004 23. Crooks CY, MD, Zumsteg JM, Bell KR. Traumatic Brain Injury: A Review of Practice Management and Recent Advances. Phys Med Rehabil Clin N Am 18: 681– 710, 2007 24. Cunningham AS, Salvador R, Coles JP, Chatfield DA, Bradley PG, Johnston AJ, Steiner LA, Fryer TD, Aigbirhio FI, Smielewski P, Williams GB, Carpenter TA, Gillard JH, Pickard JD, Menon DK. Physiological thresholds for irreversible tissue 62 damage in contusional regions following traumatic brain injury. Brain 128: 1931– 1942, 2005 25. Cuzzocrea S, Wang ZQ. Role of poly (ADP-ribose) glycohydrolase (PARG) in shock, ischemia and reperfusion. Pharmacol Res 52: 100–8, 2005 26. Cuzzocrea S. Shock, inflammation and PARP. Pharmacol Res 52: 72–82, 2005. 27. De Vries HE, Kuiper J, De Boer AG, Van Berkel TJ, Breimer DD: The blood-brain barrier in neuroinflammatory diseases. Pharmacol Rev 49: 143–155, 1997. 28. Denny-Brown D, Russell WR. Experimental cerebral concussion. J Physiol 99(1):153, 1940. 29. DeWitt DS, Prough D. Traumatic cerebral vascular injury: the effects of concussive brain injury on the cerebral vasculature. J Neurotrauma 20: 795–825, 2003 30. Di Gennaro A, Carnini C, Buccellati C, Ballerio R, Zarini S, Fumagalli F, Viappiani S, Librizzi L, Hernandez A, Murphy RC, Constantin G, De Curtis M, Folco G, Sala A. Cysteinyl-leukotrienes receptor activation in brain inflammatory reactions and cerebral edema formation: a role for transcellular biosynthesis of cysteinylleukotrienes. FASEB J 18(7): 842–844, 2004. 31. Eldadah BA, Faden AI. Caspase pathways, neuronal apoptosis, and CNS injury. J Neurotrauma 17: 811–829, 2000 32. Ergüngör MF. Kafa travmalarında patofizyoloji, Temel Nöroşirürji. Türk Nöroşirürji Derneği Yayınları. Ankara 27: 298–305. 2005 33. Fang SH, Wei EQ, Zhou Y, Wang ML, Zhang WP, Yu GL, Chu LS, Chen Z. Increased expression of cysteinyl leukotriene receptor–1 in the brain mediates neuronal damage and astrogliosis after focal cerebral ischemia in rats. Neuroscience 140(3): 969–979, 2006 34. Fiske CH, Subbarow Y. The colorimetric determination of phosphorus. J Biol Chem 66: 375–400, 1925 35. Foda MA, Marmarou A. A new model of diffuse brain injury in rats. Part II: Morphological characterization. J Neurosurg 80(2): 301–313. 1994 36. Gate L, Paul J, Ba G.N, Tew K.D, Tapiero H. Oxidative stress induced in pathologies: the role of antioxidants. Biomed Pharmacother 53: 169–180, 1999. 37. Gennarelli TA, Thibault LE, Adams JH. Diffuse axonal injury and traumatic coma in the primate. Ann Neurol 12(6): 564–574, 1982 63 38. Genovese T, Rossi A, Mazzon E, Di Paola R, Muià C, Caminiti R, Bramanti P, Sautebin L, Cuzzocrea S. Effects of zileuton and montelukast in mouse experimental spinal cord injury. Br J Pharmacol 153(3): 568–582, 2008 39. Gilgun-Sherki Y, Melamed E, Offen D. Oxidative stress inducedneurodegenerative diseases: the need for antioxidants that penetrate the blood brain barrier. Neuropharmacology 40: 959–975, 2001 40. Glenn TC, Kelly DF, Boscardin WJ, McArthur DL, Vespa P, Oertel M, Hovda DA, Bergsneider M, Hillered L, Martin NA. Energy dysfunction as a predictor of outcome after moderate or severe head injury: indices of oxygen, glucose, and lactate metabolism. J Cereb Blood Flow Metab 23: 1239–1250, 2003 41. Graham DI, McIntosh TK, Maxwell WL, Nicoll JA. Recent advances in neurotrauma, J Neuropathol Exp Neurol 59 641– 651, 2000 42. Graham DI, Adams JH, Doyle D, Teasdale GM, Lawrence AE, McLellan DR. Ischaemic brain damage is still common in fatal non-missile head injury. J Neurol Neurosurg Psychiatry 52: 346–350, 1989 43. Graham DI, Neuropathology of the head injury. In: Narayan RK, Wilberger JE, Povllishock JT. eds. Neurotrauma. Mc Graww Hill: New York; p.43–58, 1996 44. Greenberg MS, Handbook of Neurosurgery. Sixth Ed. Florida: Thieme p.632–637, 2006 45. Hall ED, Braughjer JM. Central nervous system and stroke. II. Physiological and pharmacological evidence for involvement of oxygen radicals and lipid peroxidation. Free Radic Biol Med 6: 303–313, 1989 46. Hausmann R, Biermann T, Wiest I, Tübel J, Betz P. Neuronal apoptosis following human brain injury. Int J Legal Med 118(1): 32–36, 2004. 47. Hillegass LM, Griswold DE, Brickson B, Albrightson-Winslow C. Assessment of myeloperoxidase activity in whole rat kidney. J Pharmacol Methods 24: 285–295, 1990 48. Hu H, Chen G, Zhang JM, Zhang WP, Zhang L, Ge QF, Yao HT, Ding W, Chen Z, Wei EQ. Distribution of cysteinyl leukotriene receptor 2 in human traumatic brain injury and brain tumors. Acta Pharmacol Sin 26: 685–690, 2005 49. Huang XJ, Zhang WP, Li CT, Shi WZ, Fang SH, Lu YB, Chen Z, Wei EQ. Activation of CysLT receptors induces astrocyte proliferation and death after oxygen-glucose deprivation Glia 56(1): 27–37, 2008 64 50. Iacoangeli M, Roselli R, Pompucci A, Scerrati M. Acute menagement of head injury. Contemp Neurosurg 22: l–8, 2000 51. Inoue Y, Shiozaki T, Tasaki O, Hayakata T, Ikegawa H, Yoshiya K, Fujinaka T, Tanaka H, Shimazu T, Sugimoto H. Changes in cerebral blood flow from the acute to the chronic phase of severe head injury. J Neurotrauma 22: 1411–1418, 2005 52. Jennett B, Bond M. Assessment of outcome after severe brain damage. A practical scale. Lancet 1: 480–484, 1975 53. Jennett B, Historical perspective on head injury. In: Narayan RK, Wilberger JE, Povllishock JT. eds. Neurotrauma. Mc Graww Hill: New York; p.3–12, 1996 54. Johnston AJ, Steiner LA, Coles JP, Chatfield DA, Fryer TD, Smielewski P, Hutchinson PJ, O'Connell MT, Al-Rawi PG, Aigbirihio FI, Clark JC, Pickard JD, Gupta AK, Menon DK. Effect of cerebral perfusion pressure augmentation on regional oxygenation and metabolism after head injury. Crit Care Med 33: 189–195, 2005 55. Jones TR, Labelle M, Belley M, Champion E, Charette L, Evans J, Ford-Hutchinson AW, Gauthier JY, Lord A, Masson P, McAuliffe M, McFarlane CS, Metters KM, Pickett C, Piechuta H, Rochette C, Sawyer R, Rodger IW, Yoyng RN, Zamboni R, Abraham WM. Pharmacology of montelukast sodium (Singulair), a potent and selective leukotriene D4 receptor antagonist. Can J Physiol Pharmacol. 73(2): 191– 201, 1995 56. Kelley BJ, Lifshitz J, Povlishock JT. Neuroinflammatory responses after experimental diffuse traumatic brain injury. J Neuropathol Exp Neurol 66(11): 989–1001, 2007 57. Kelly DF, Kordestani RK, Martin NA, Nguyen T, Hovda DA, Bergsneider M, McArthur DL, Becker DP. Hyperemia following traumatic brain injury: relationship to intracranial hypertension and outcome. J Neurosurg 85 (5): 762–771, 1996 58. Kelly DF, Martin NA, Kordestani R, Counelis G, Hovda DA, Bergsneider M, McBride DQ, Shalmon E, Herman D, Becker DP. Cerebral blood flow as a predictor of outcome following traumatic brain injury. J Neurosurg 86 (4): 633–641, 1997 59. Kempski O. Cerebral edema. Semin Nephrol 21(3): 303–307, 2001 60. Kraus JF, McArthur DL, Silverman TA, Jarayaman M, Epidemiology of brain injury In: Narayan RK, Wilberger JE, Povllishock JT. eds. Neurotrauma. Mc Graww Hill: New York; p.13–30, 1996 61. Lee JH, Kelly DF, Oertel M, McArthur DL, Glenn TC, Vespa P, Boscardin WJ, Martin NA. Carbon dioxide reactivity, pressure autoregulation, and metabolic 65 suppression reactivity after head injury: a transcranial Doppler study. J Neurosurg 95: 222–232, 2001 62. Lima FD, Souza MA, Furian AF, Rambo LM, Ribeiro LR, Martignoni FV, Hoffmann MS, Fighera MR, Royes LF, Oliveira MS, de Mello CF. Na+, K+-ATPase activity impairment after experimental traumatic brain injury: relationship to spatial learning deficits and oxidative stress. Behav Brain Res 193(2): 306–310, 2008 63. Lowry OH, Rosenbrough NJ, Farr AL, Randall RJ. Protein measurements with the folin phenol reagent. J Biol Chem 193: 265-75, 1951. 64. Lucas SM, Rothwell NJ, Gibson RM. The role of inflammation in CNS injury and disease. Br J Pharmacol 147 Suppl 1: 232–240, 2006 65. Maas AI, Dearden M, Servadei F, Stocchetti N, Unterberg A. Current recommendations for neurotrauma. Curr Opin Crit Care 6 (74): 281– 292, 2000 66. Magnoni S, Ghisoni L, Locatelli M, Caimi M, Colombo A, Valeriani V, Stocchetti N. Lack of improvement in cerebral metabolism after hyperoxia in severe head injury: a microdialysis study. J Neurosurg 98: 952–958, 2003 67. Marmarou A, Foda MA, van den Brink W, Campbell J, Kita H, Demetriadou K. A new model of diffuse brain injury in rats. Part I: Pathophysiology and biomechanics. J Neurosurg 80(2): 291–300, 1994 68. Marmarou A. A review of progress in understanding the pathophysiology and treatment of brain edema. Neurosurg Focus 22(5): E1, 2007 69. Marshall LF. Head injury: recent past, present, and future. Neurosurgery 47(3): 546– 561, 2000 70. Martin NA, Patwardhan RV, Alexander MJ, Africk CZ, Lee JH, Shalmon E, Hovda DA, Becker DP. Characterization of cerebral hemodynamic phases following severe head trauma: hypoperfusion, hyperemia, and vasospasm. J Neurosurg 87: 9–19, 1997 71. Miller JD, Piper IR, Jones PA, Pathophysiology of head injury. In: Narayan RK, Wilberger JE, Povllishock JT. eds. Neurotrauma. Mc Graww Hill: New York; p.61– 69, 1996. 72. Miller DC. Use of perfusion fixation for improved neuropathologic examination. Arch Pathol Lab Med. 122(11):949, 1998. 73. Moos MP, Mewburn JD, Kan FW, Ishii S, Abe M, Sakimura K, Noguchi K, Shimizu T, Funk CD. Cysteinyl leukotriene 2 receptor-mediated vascular permeability via transendothelial vesicle transport. FASEB J 22(12): 4352–4362, 2008 66 74. Morganti-Kossmann MC, Satgunaseelan L, Bye N, Kossmann T. Modulation of immune response by head injury. Injury 38(12): 1392–1400, 2007 75. Obrenovitch TP, Urenjak J. Is high extracellular glutamate the key to excitotoxicity in traumatic brain injury? J Neurotrauma 14: 677–698, 1997 76. Oliver I. Schmidt, Christoph E. Heyde, Wolfgang Ertel, Philip F. Stahel. Closed head injury—an inflammatory disease? Brain Research Reviews 48: 388– 399, 2005 77. Overgaard J, Tweed WA. Cerebral circulation after head injury. J Neurosurg 59: 439– 446, 1983 78. Park Park E, Bell JD, Baker AJ. Traumatic brain injury: can the consequences be stopped? CMAJ 178(9): 1163–1170, 2008 79. Potts MB, Koh SE, Whetstone WD, Walker BA, Yoneyama T, Claus CP, Manvelyan HM, Noble-Haeusslein LJ. Traumatic injury to the immature brain: inflammation, oxidative injury, and ironmediated damage as potential therapeutic targets. NeuroRx 3: 143–153, 2006 80. Praticò D, Reiss P, Tang LX, Sung S, Rokach J, McIntosh TK. Local and systemic increase in lipid peroxidation after moderate experimental traumatic brain injury. J Neurochem 80(5): 894–898, 2002 81. Reading HW, Isbir T. The role of cation activated ATPase in transmitter release from the art iris. Q J Exp Physiol Cogn Med Sci 65: 105–116, 1980 82. Reilly PL. Brain injury: the pathophysiology of the first hours. 'Talk and Die revisited'. J Clin Neurosci 8(5): 398–403, 2001 83. Robertson CL, Bell MJ, Kochanek PM, Adelson PD, Ruppel RA, Carcillo JA, Wisniewski SR, Mi Z, Janesko KL, Clark RS, Marion DW, Graham SH, Jackson EK. Increased adenosine in cerebrospinal fluid after severe traumatic brain injury in infants and children: association with severity of injury and excitotoxicity. Crit Care Med 29: 2287–3393, 2001 84. Rodriguez-Baeza A, Reina-De La Torre F, Poca A, Marti M, Garnacho A. Morphological features in human cortical brain microvessels after head injury: a threedimensional and immunocytochemical study. Anat Rec Part A 273: 583–593, 2003 85. Rovati GE Capra V. Cysteinyl-leukotriene receptors and cellular signals. ScientificWorldJournal. 7, 185: 1375–1392, 2007 86. Royo NC, Shimizu S, Schouten JW, Stover JF, McIntosh TK. Pharmacology of traumatic brain injury. Curr Opin Pharmacol 3(1): 27–32, 2003 67 87. Sala A, Folco G. Neutrophils, endothelial cells, and cysteinyl leukotrienes: a new approach to neutrophil-dependent inflammation? Biochem Biophys Res Commun 283(5): 1003–1006, 2001 88. Schoettle R.J, Kochanek P.M, Magargee M.J, Uhl M.W, Nemoto E.M, Early polymorphonuclear leukocyte accumulation correlates with the development of posttraumatic cerebral edema in rats, J Neurotrauma 7: 207– 217, 1990 89. Schuhmann MU, Mokhtarzadeh M, Stichtenoth DO, Skardelly M, Klinge PM, Gutzki FM, Samii M, Brinker T. Temporal profiles of cerebrospinal fluid leukotrienes, brain edema and inflammatory response following experimental brain injury. Neurol Res 25(5): 481–491, 2003 90. Sener G, Sehirli O, Toklu H, Ercan F, Alican I. Montelukast reduces ischaemia/ reperfusion-induced bladder dysfunction and oxidant damage in the rat. J Pharm Pharmacol 59(6): 837–842, 2007 91. Sener G, Sehirli O, Velioğlu-Oğünç A, Cetinel S, Gedik N, Caner M, Sakarcan A, Yeğen BC. Montelukast protects against renal ischemia/reperfusion injury in rats. Pharmacol Res 54(1): 65–71, 2006 92. Shao CX, Roberts KN, Markesbery WR, Scheff SW, Lovell MA. Oxidative stress in head trauma in aging. Free Radic Biol Med 41: 77–85, 2006 93. Sheng WW, Li CT, Zhang WP, Yuan YM, Hu H, Fang SH, Zhang L, Wei EQ. Distinct roles of CysLT1 and CysLT2 receptors in oxygen glucose deprivationinduced PC12 cell death. Biochem Biophys Res Commun 346(1): 19–25, 2006 94. Slemmer JE, Shacka JJ, Sweeney MI, Weber JT. Antioxidants and free radical scavengers for the treatment of stroke, traumatic brain injury and aging. Curr Med Chem 15(4): 404–414, 2008 95. Smith QR. Transport of glutamate and other amino acids at the blood-brain barrier. J Nutr 130 (4S Suppl): 1016–1022, 2000 96. Stein SC, Clasification of head injury. In: Narayan RK, Wilberger JE, Povllishock JT. eds. Neurotrauma. Mc Graww Hill: New York; p.31–41, 1996 97. Stiefel MF, Udoetuk JD, Spiotta AM, Gracias VH, Goldberg A, Maloney-Wilensky E, Bloom S, Le Roux PD. Conventional neurocritical care and cerebral oxygenation after traumatic brain injury. J Neurosurg 105: 568–575, 2006 98. Tavazzi B, Signoretti S, Lazzarino G, Amorini AM, Delfini R, Cimatti M, Marmarou A, Vagnozzi R. Cerebral oxidative stress and depression of energy metabolism correlate with severity of diffuse brain injury in rats. Neurosurgery 56: 582–589, 2005 68 99. Teasdale G, Jennett B. Assessment of coma and impaired consciousness. A practical scale. Lancet 2(7872): 81–84, 1974 100. Toklu HZ. Doktora tezi. Danışmanlar: Prof. Dr. Meral Keyer Uysal, Doç. Dr. Levent Kabasakal. Sıçanlarda sepsise bağlı ensefalopatide erken ve geç dönemde riluzolün etkilerinin araştırılması [The effects of riluzole in early and late phase of sepsis-induced encephalopaty in rats] Yapıldığı Yer: Marmara Üniversitesi · Sağlık Bilimleri Enstitüsü · · Farmakoloji Anabilim Dalı . YÖK Tez no: 193720, 2006. 101. Ucar T, Tanriover G, Gurer I, Onal MZ, Kazan S. Modified experimental mild traumatic brain injury model. J Trauma 60(3): 558–565, 2006 102. Unterberg AW, Stover J, Kress B, Kiening KL. Edema and brain trauma. Neuroscience. 129(4): 1021–1029, 2004 103. Verweij BH, Muizelaar JP, Vinas F, Peterson PL, Xiong Y, Lee CP. Impaired cerebral mitochondrial function after traumatic brain injury in humans. J Neurosurg. 93: 815–820, 2000 104. Wang ML, Huang XJ, Fang SH, Yuan YM, Zhang WP, Lu YB, Ding Q, Wei EQ. Leukotriene D4 induces brain edema and enhances CysLT2 receptor-mediated aquaporin 4 expression. Biochem Biophys Res Commun 350(2): 399–404, 2006 105. Werner C, Engelhard K. Pathophysiology of traumatic brain injury. Br J Anaesth 99(1): 4–9, 2007 106. Wu G, Fang Y.Z, Yang S, Lupton J.R, Turner N.D: Glutathione metabolism and its implications for health. J. Nutr, 134: 489–492, 2004 107. Yu GL, Wei EQ, Zhang SH, Xu HM, Chu LS, Zhang WP, Zhang Q, Chen Z, Mei RH, Zhao MH.Montelukast, a cysteinyl leukotriene receptor–1 antagonist, doseand time-dependently protects against focal cerebral ischemia in mice. Pharmacology 73(1): 31–40, 2005 108. Zhang WP, Hu H, Zhang L, Ding W, Yao HT, Chen KD, Sheng WW, Chen Z, Wei EQ. Expression of cysteinyl leukotriene receptor 1 in human traumatic brain injury and brain tumors. Neurosci Lett 363(3): 247–251, 2004 109. Zhang Z, Artelt M, Burnet M, Trautmann K, Schluesener HJ. Early infiltration of CD8 macrophages/microglia to lesions of rat traumatic brain injury. Neuroscience 141: 637–644, 2006 110. Zhuang J, Shackford S.R, Schmoker J.D, Anderson ML. The association of leukocytes with secondary brain injury. J Trauma 35: 415– 422, 1993 69 8. Etik Kurul Raporu 70