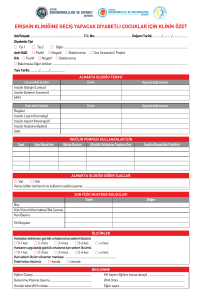
diabetes mellitus`un tanımı, tanısı ve sınıflaması
advertisement

T.C. SAĞLIK BAKANLIĞI ŞİŞLİ ETFAL EĞİTİM VE ARAŞTIRMA HASTANESİ Şef. Doç. Dr. Yüksel ALTUNTAŞ YENİ TESPİT TİP 2 DİABETES MELLİTUSLU HASTALARDA PANKREAS BETA HÜCRE REZERVİNİN DEĞERLENDİRİLMESİ (Umanlık Tezi) Dr. Güliz Serin Yalçın İstanbul - 2004 1 Önsöz Asistanlığım süresince tıbbi tecrübe ve bilgisi yanında sosyokültürel yönünden de yararlandığım, yeniliklere ve değişikliğe açık, yönetici ve eğitici olarak her zaman saygı duyduğum değerli hocam Doç. Dr. Yüksel Altuntaş’a şükranlarımı sunuyorum. Kısa bir süre de olsa birlikte çalışma zevkini tattığım Hematolog Dr. Dilek Argon’a, bana Hematoloji bilimini sevdirdiği ve hekimlik hayatım boyunca hastalarla kuracağım iletişimde örnek teşkil ettiği için teşekkürlerimi sunuyorum. Rotasyonlarım sırasında birlikte çalışmak şerfefine eriştiğim Şişli Etfal Eğitim ve Araştırma Hastanesi Nefroloji Kliniği Şefi Dr. Abdülkadir Ünsal’a, Gastroenteroloji Kliniği Şefi dr. Mehmet Sökmen’e ve şef yardımcısı dr. Nihat Akbayır’a teşekkür ederim. Birlikte çalıştığım servis uzmanlarım, asistan arkadaşlarım, hemşire ve personele teşekkür ederim. Rotasyonlarım sırasında birlikte çalıştığım sayın hocalarım; doç. Dr. Filiz Koşar (S.B. Yedikule Göğüs Hastalıkları Hastanesi 6. Klinik Şefi), Dr Nezaket Eren (S. B. Şişli Etfal Eğitim ve Araştırma Hastanesi Biyokimya Kliniği şefi), Dr. Engin Seber (S. B. Şişli Etfal Eğitim ve Araştırma Hastanesi Enfeksiyon Hastalıkları Kliniği emekli şefi), Koşuyolu Göğüs Kalp Damar Cerrahisi Hastanesi Kardiyoloji bölümünün değerli hocalarına ayrı ayrı teşekkür ederim. Bugünlere gelmem için benden hiçbir fedakarlığı esirgemeyen sevgili anneme ve babama, tezimin yazılma aşamasında çok büyük katkısı olan sevgili eşim Selçuk Yalçın’a, manevi desteğini her zaman yanımda hissettiğim sevgili kardeşim Mert Serin’e en içten duygularımla teşekkür ederim. Dr Güliz Yalçın Serin 2 İÇİNDEKİLER GİRİŞ 1-2 GENEL BİLGİLER 3-37 MATERYAL VE METOD 38-40 BULGULAR 41-47 TARTIŞMA VE SONUÇ 48-55 KAYNAKLAR 56-63 3 GİRİŞ Tip 2 diyabetli hastalarda insülin tedavisi ile ilgili olarak uygulanan genel yaklaşım, önce bir oral antidiyabetik kullanıldıktan sonra bu ilaçlara karşı sekonder yetersizlik geliştikten sonra insülin tedavisine başlamak şeklindedir. Beta hücre fonksiyonu endojen insülinin pulsatil salınım göstermesi ve kısa yarı ömre sahip olması (6-7 dk.) nedeniyle, endojen insülin düzeylerine bakılarak sağlıklı bir şekilde değerlendirilemez. Beta hücre fonksiyonunu değerlendirmede daha uygun bir yöntem endojen insülin üretiminin bir başka göstergesi olan ve daha uzun yarı ömre sahip (30 dk.) C-peptid düzeylerine bakılmasıdır. Bununla birlikte düşük C-peptid düzeyi bulunması, hastada beta hücre fonksiyonu kaybı mı, yoksa geri dönüşümlü glikoz toksisitesi mi olduğunu ayırt etmede yeterince yardımcı olmaz. Hipergliseminin kendisi hem beta hücresi üzerine etki ederek insülin salgılanmasını baskılar hem de periferik dokularda insülinin kullanılmasını azaltır. Hipergliseminin beta hücresi üzerine olan bu olumsuz etkisine glukoz toksisitesi adı verilmektedir. Hiperglisemi durumunda sıkı metabolik kontrol ile (diyet, sulfonilüre ve insülin tedavisi ile) insülin salgılanmasının düzeldiğinin gözlenmesi hipergliseminin kendisinin insülin salgılanması üzerine baskılayıcı bir etkisinin olabileceğini düşündürmüştür. Diyabetik hastalarda, herhangi bir tedavinin insülin sekresyonu üzerine olan etkisi konusunda hükme varmak için, uygulanan testlerin güvenilir olması ve standardazisyonunun uygun olması gerekmektedir. C-peptid-Glukagon testi insülin sekresyonunu belirlemek amacıyla kullanılan testler arasında en standardize testtir. Uzun yıllardır bir çok açıdan geçerliliği kanıtlanmıştır. C-peptid-Glukagon testi, sülfonilüreler veya diğer antidiyabetik ilaçlarla 4 sürdürülen tedaviler esnasında gerçekleştirilir. Bu ilaçlar, aynı zamanda endojen insülin sekresyonunu da etkilerler. Sülfonilüreler ile tedavi edilen hastaların her yıl %5-10’unda sulfonilüre yetersizliği gelişmektedir. Proinsülin insülinin ancak %5’i kadar biyolojik etkiye sahip olup insülin immünoreaktivitesinin normal bireylerde %2-4’ünü, NIDDM’lu bireylerde ise %8-10’unu oluşturur. Proinsülinin %70’ini 32-33 split (kırılmış) proinsülin oluşturur Proinsülin ve split proinsülinlerin klirensleri yavaş olduğundan ve de insülin ölçümünde kullanılan rutin RIA yöntemleri insülinin yanında proinsülinleri de (sağlam ve kırılmış) ölçtüğünden insülin düzeyleri olduğundan yüksek bulunur. Buradan yola çıkarak plazmadaki sağlam ve 32-33 kırılmış proinsülin konsantrasyonlarının ölçümünün (çift işaretli immünometrik yöntemler ile) insülin direncine veya beta hücre salgılama kapasitesine ya da her ikisine bağlı olarak beta hücresinde oluşan fonksiyon bozukluğunu yansıtabileceği ileri sürülmektedir. Daha önce yapılan çalışmalarda kısa etkili insülinler ve NPH insülin ile yapılan intensif insülin tedavisinin Beta-hücre rezervini iyileştirdiği gösterilmiştir.Tip 2 diabette insülin analogları ile daha fizyolojik bir tedavi yaklaşımı sağlanmaktadır. Tip 2 diabette 1. faz insülin sekresyonu bozulmuştur.1. faz insülin yanıtı prandial glukoz toleransının en önemli belirleyicisidir. Hızlı etkili insülin analogları postprandial hiperglisemiyi 1.faz insülin yanıtını düzelterek kısa etkili insülinlere göre daha iyi düşürmektedir. Bu çalışmada bizim amacımız yeni tip2 diabetes mellitus tanısı almış olan hastalarda 3’lü insülin analogları ve NPH insülin ile uygulanan intensif insülin tedavisinin Beta-hücre rezervi üzerine olan etkisini belirlemektir. 5 6 GENEL BİLGİLER DİABETES MELLİTUS’UN TANIMI, TANISI VE SINIFLAMASI(1) DİABETES MELLİTUS’UN TANIMI Diabetes Mellitus insülin hormon sekresyonunun ve/veya insülin etkisinin mutlak veya göreceli azlığı sonucu karbonhidrat, protein ve yağ metabolizmasında bozukluklara yol açan kronik hiperglisemik bir grup metabolizma hastalığıdır. Diabetes Mellitus klinik olarak polidipsi, poliüri, polifaji, pruritus, ağırlık kaybı gibi klasik belirtiler ve hastalığa spesifik retinopati, nöropati, nefropati gibi komplikasyonlar ile şüphe edilebilir veya tanınabilir. Özellikle insüline bağımlı olmayan diabet bu belirtileri göstermiyebilir, böyle durumlarda tanı kan ve idrar testine göre konur. İnsüline bağımlı olan diabet ise klasik belirtiler ile hemen tanınır. DİABETES MELLİTUS’UN VE BOZULMUŞ GLUKOZ TOLERANSININ TANISI Daha önceki yapılan çalışmalarda diabetin spesifik ve en yaygın komplikasyonlarından olan retinopati oluşumunun glukoz yüklemesinden sonraki 2. saatteki 200 mg/dl plazma glukoz düzeyi ile yakın ilişkili olduğu bunun da 140 mg/dl açlık plazma glukozuna karşılık geldiği gösterilmişti. Fakat bu konuda yapılan yeni çalışmalarda bunun doğru olmadığı retinopati gelişme açısından gerçekte 120 ve 126 mg/dl arasındaki açlık plazma glukoz düzeylerinin glukoz yüklemesinden sonraki 2. saatteki 200 mg/dl plazma glukoz düzeyleri ile ilişkili olduğu ileri sürülmüştür. Diabetes Mellitus’un yeni tanı kriterleri tablo 1’ de gösterilmiştir. 7 Amerikan Diabet Birliğine (ADA) göre diabetes mellitus’ un en basit tanısı açlık gliseminin venöz plazmada en az iki ardışık ölçümde 126 mg/dl veya daha yüksek olması ile konur. Yine günün herhangi bir saatinde açlık ve tokluk durumuna bakılmaksızın randomize venöz plazma glisemisinin 200 mg/dl’ in üzerinde olması ve polidipsi, poliüri, polifaji, zayıflama gibi diabetik semptomlarının oluşu ile de tanı konulabilir . Tablo 1: Diabetes Mellitusun Tanı Kriterleri 1. Diabet semptomları ve 200 mg/dl randomize plazma glukoz düzeyi: Günün herhangi bir saatinde öğüne bakılmaksızın ölçülen plazma glisemi değeri Poliüri Polidipsi Açıklanamayan ağırlık kaybı 2. Açlık plazma glukoz düzeyi 126 mg/dl: En az 8 saatlik tam açlık sonrası 3. Oral glukoz tolerans testi sırasında 2.saat plazma glukoz düzeyi 200 mg/dl Açlık plazma glukoz düzeyi 110 mg/dl altında olan ve diabet açısından yüksek risk taşıyan bireylerde belirli aralıklarla OGTT yapılarak bozulmuş glikoz toleransı veya diabet aranmalıdır(tablo 2). Açlık kan şekeri tek başına tanı kriterini sağlıyorsa OGTT’ ne gerek yoktur. Eğer hastada semptomlar yok veya hafif var ise ve glisemi tanı sınırlarını zorluyor ise OGTT gerekebilir. Ayrıca bozulmuş glukoz tolerans tanısı için de OGTT’ ne gerek vardır. Tip 1 diabet tanısı için OGTT’ ne gerek yoktur. Aslında tablo 2 de gösterilen ve Amerikan Diabet Birliği (ADA) tarafından 1997’ de önerilen yeni tanı kriterleri OGTT yapılmadan da açlık plazma glukozuna göre tanı konulmamış büyük bir hasta popülasyonunun tanısını kolaylaştırmaktadır. Tablo 2: Glukoz Toleransının Sınıflaması (ADA 1997) Açlık Plazma Glukozu Normal < 110 mg/dl Bozulmuş açlık glukozu 110 mg/dl ve < 126 mg/dl Diabet 126 mg/dl 8 OGTT sırasında 2. Saat plazma glukozu Normal < 140 mg/dl Bozulmuş glukoz toleransı 140 ve < 200 mg/dl Diabet 200 mg/dl Tablo 3: Diabet Açısından Yüksek Risk Grupları (WHO 1994) Tip-2 diabetiklerin birinci dereceden akrabaları Ailede genetik yüklülük (Ailede yoğun Tip-2 diabetli varlığı). Kırsal alandan kentsel alana göç edenler veya aktif bir yaşamdan pasif bir yaşama dönmüş kişiler. Beden kitle indeksi 27 kg/m2 üzeri olan ve bel/kalça oranı 1.0’ den büyük olan obez ve/veya android obezler Daha önce gestasyonel diabet saptanmış olan veya iri bebek doğan kadınlar (>4 kg bebek). Metabolik (sendrom X) sendromlu kişiler Sekonder diabete yol açabilecek hastalığı olanlar Diabetojenik ilaç kullananlar Glikozürisi bulunan kişiler ADA açlık plazma glukoz düzeyinde bir değişiklik yaparak 140 mg/dl yerine 126 mg/dl’ lik glisemi düzeyini kabul etmiş ve 110 mg/dl ile 126 mg/dl arasındaki değer için bozulmuş açlık glukozu adını verdiği yeni bir tanımlama önermiştir. DİABETES MELLİTUSUN SINIFLAMASI İlk kez 1979 yılında NDDG daha sonra da 1985 yılında WHO tarafından diabetin geniş bir sınıflaması yapılmıştır.WHO’ nun yaptığı sınıflama kliniksel olup aynı zamanda diabeti terminolojik olarak insüline bağımlı (IDDM) ve insüline bağımlı olmayan (NIDDM) olarak da adlandırmıştı. IDDM ve NIDDM uygulanabilirliği sınırlıdır. heterojen olduğundan WHO sınıflamasının genel Buna karşın, WHO ve NDDG sınıflamaları, epidemiyolojik 9 çalışmalarda ve aynı derecede hastaların tedavisinde klinik araştırma ve terapötik ayrımı için önemli ve gerekli yönergeleri sağlamıştır. Diabet heterojenitesiyle ilgili en önemli güncel konu IDDM ve NIDDM arasındaki ve kendi içlerindeki olası etyoloji ve fenotipik farklılıklardır. Daha sonra ADA tarafından 1998 yılında önerilen yeni sınıflama ise etyolojik olup keza insüline bağımlı ve insüline bağımlı olmayan diabet yerine tip 1 ve tip 2 diabet terminolojisini de önermektedir(tablo 4). Tablo 4: Diabetes Mellitus’un Etyolojik Sınıflaması (ADA 1997) I-Tip 1 diabetes (B hücre yıkımı, çoğunlukla mutlak insülin eksikliği) A- İmmunolojik B- İdiopatik II-Tip 2 diabetes İnsülin direnci veya insülin salgı bozukluğu ağırlıklı olarak neden olabilir. III-Diğer spesifik tipler A- B hücre fonksiyonunda genetik defekt 1-Kromozom 12 ,HNF-1 alfa (MODY 3) 2-Kromozom 7,glukokinaz (MODY 2) 3-Kromozom 20,HNF-4 alfa(MODY 1) 4- Mitokondriyal DNA 5-Diğerleri B- İnsülin etkisinde genetik defekt 1-Tip A insülin resistansı 2-Leprechaunizm 3-Rabson-Mendenhall sendromu 4-Lipoatrophic diabet 5-Diğerleri C- Ekzokrin pankreas hastalıkları 1-Pankreatit 2-Travma/pankreatektomi 3-Neoplazm 4-Kistik fibrosis 5-Hemakromatozis 6-Fibrokalküloz pankreas 7-Diğerleri D- Endokrinopati 1-Akromegali 2-Cushing sendromu 3-Glukagonoma 4-Feokromasitoma 5-Hipertiroidizm 6-Somatostatinoma 7-Aldesteronoma 8-Diğerleri E- İlaç yada kimyasallara bağlı 1-Vacor 2-Pentamidin 3-Nikotinik asit 4-Glukokortikoidler 5-Tiroid hormonu 6-Diazoksit 7-B-adrenerjik agonistler 8-Tiazidler 9-Dilantin 10 10-Alfa-interferon 11-Diğerleri F- Enfeksiyonlar 1-Konjenital rubella 2-Sitomegalovirus 3-Diğerleri G- İmmun Diabetin bilinmeyen formları 1-“Stiff-man” sendromu 2-Anti-insülin antikoru 3-Diğerleri H- Diabetle bazen birlikteliği olan genetik sendromlar 1-Down sendromu 2-Klinefelter sendromu 3-Turner sendromu 4-Wolfram sendromu 5-Friedreich ataksisi 6-Huntington korea 7-Laurence-Moon-Biedl sendromu 8-Miyotonik distrofi 9- Porfiria 10-Prader-Willi sendromu 11-Diğerleri Tip 2 diabet hastaların önemli bir bölümü tipik olarak obez ve hiperinsülinemik iken, insülin hiposekresyonunun görüldüğü obez olmayan Tip 2 diabet hastaları da bulunmaktadır ve tanıyı izleyen birkaç ay veya yıl içinde oral hipoglisemik ilaçlarla yapılan tedavi başarısız olmakta, bunun sonucunda da insülin bağımlılığına doğru bir ilerleme olmaktadır. Diğer bir deyişle klasik Tip 1 diabet ve Tip 2 diabet, sadece diğer bir sınıflandırma ölçeğinin zıt kutuplarıdır. Bunun bir göstergesi olarak Tip 2 diabet grubundaki bazı kişiler ilk klinik prezentasyonda Tip 2 diabet olarak tanınırlar fakat gerçekte yavaş ilerleyen bir Tip 1 diabet hastasıdırlar. Bu durum son yıllarda yavaş seyirli tip 1 diabet veya yetişkinlerin Latent Otoimmün Diabeti (LADA) olarak bilinmektedir. Diabetin sınıflandırılmasındaki önemli bir değişiklik, 1985 WHO Çalışma Grubu’unca önerilen, daha önceleri tropikal diabet olarak adlandırılan ve Tip 1 diabet ve Tip 2 diabet ile sıralamaya giren ana bir klinik alt tip olan Malnütrisyona Bağlı Diabetes Mellitus’ un (MRDM) ortaya çıkması oldu. MRDM, ne Tip 1 diabet ne de Tip 2 diabet kategorilerine tam anlamıyla girmektedir ve başka bir terminoloji olan fibrokalkülöz pankreatik diabet de yaygın olarak kullanılmaktadır. Çoğunlukla genç yetişkinleri etkileyen bu hastalık, şiddetli ketoasidozisle birlikte olmadığı ve insülin bağımlılığı da aralıklı olduğundan “Fazik İnsüline Bağımlı Diabetes 11 Mellitus” (PIDDM) olarakta tanımlanmaktadır. Hindistan, Bangladeş ve Endonezya’ nın belli bölgelerini de içine alan bazı ülkelerde MRDM’ nin batı ülkelerinde Tip 2 diabet’ nin olduğu kadar yaygın olduğu görüşü ileri sürülmektedir, fakat destekleyici epidemiyolojik veriler yetersizdir. Kliniksel yapılan sınıflama diabetes mellitus, bozulmuş glikoz toleransı ve gestasyonel diabet olmak üzere 3 grup olup hepsinde mevcut olan aşikar hiperglisemi tedavi gerektirmektedir. Preklinik dönemdeki diabette ise hiperglisemi bulunmamaktadır. Tip-1 diabetes mellitus’ a uyan HLA antijenlerine sahip olup, normoglisemisi olan ve dolaşımda islet hücresi antikorları veya insüline karşı otoantikorlar saptanan bireylerde olarak büyük olasılıkla diabetes mellitus geçirecekler arasındadır. İstatistiksel olarak risk altında bulunan gruba giren bireylerin erken tanı açısından periodik kontrollerden geçmesi gerekir. Klinik grubun en sık görülen tipi diabetes mellitustur. Bunlar insüline bağımlı diabet,insüline bağımlı olmayan diabet, malnütrisyonla ilgili diabet ve belirli sendromlarla ilişkili diabet olmak üzere 4 alt grupta incelenecektir. İNSÜLİNE BAĞIMLI OLMAYAN DİABETES MELLİTUS (NIDDM) Toplumda en sık görülen diabetes melllitus tipidir. İnsuline bağımlı olmayan diabetes mellitus (NIDDM) ya da tip 2 diabet polidipsi, poliüri, polifaji, pruritus, ağırlık kaybı gibi klasik belirtiler ile ortaya çıkarsa da çoğu kez uzun sürebilen asemptomatik dönemi mevcuttur. Genellikle 45 yaş üzerinde ilk yakınmalar başlar. Polidipsi, poliüri ve polifaji gibi yakınmalardan ziyade retinopati, nefropati, nöropati ve aterosklerotik kalp hastalığı gibi kronik komplikasyonlarla ilgili yakınmalar hastayı hekime ilk kez getirebilir ve çoğunlukla ilk tanı konulduğunda kronik komplikasyonlar vardır. 12 Hiperglisemiye rağmen kan ve idrarda keton cisimleri azdır veya yoktur. İnsulin tedavisi çoğu kez gerekli değildir. Ketoasidoz spontan olarak oluşmaz. Sadece aşırı hiperglisemi ve hiperozmolarite durumlarında nadiren ketoasidoz koması gelişebilir. Diabetik ketoasidoz koması, şiddetli enfeksiyon veya mezenter arter embolisi gibi acil bir durum olmadıkça gelişmez. Bu hastalarda daha sık görülebilen koma, yeterli sıvı alınmamasına bağlı gelişen hiperglisemik hiperosmolar non-ketotik komadır. Diabetik hipergliseminin patogenezinde üç önemli faktörün rol oynadığı bilinmektedir. Bunlar beta hücre insulin salgısının bozulması, insulin direnci ve karaciğerde glukoz üretiminin artışıdır. Hem insulin direnci hemde bozulmuş insulin sekresyonu tip 2 diabetin patogenezinde genetik olarak kontrol edilen faktörler olup bunlardan hangisinin primer ağırlıkta rol oynadığı henüz açık değildir. Aile öyküsü hemen hepsinde olmasına karşın hastalık henüz tek bir genetik zemine oturtulamamıştır. Yine de tip 2 diabetin çoğu formları genetik yüklülük ile ilişkilidir. Tip 2 diabet keza yaygın olarak obezite ile çok yakın ilişkilidir. Obezite insulin direncini artırarak hiperglisemiyi ağırlaştırmasına rağmen obezite olmadan da Tip 2 diabet gelişir. Bu yüzden obez ve non obez NIDMM ayırımı etiyolojik bir farklılık oluşturur. Buna göre obez Tip 2 diabet’ de insulin direnci daha önemli iken, non obez Tip 2 diabet’ de insulin sekresyon bozukluğu ön plana geçer. İnsülin rezistansı sendromu, diğer adı ile “Sendrom X”, Reaven ve arkadaşları tarafından yakın zamanda tanımlanmıştır. Bu sendromdaki primer bozukluk, periferik insülin direncidir ve diğer bulgulardan önce ortaya çıkar. Bunu obesite, aterosklerozis, hiperlipidemi ve hipertansiyon izler. Hiperinsülinizmin özellikle trunkal obesite ile ilişkisi vardır. İleri sürülen hipotezde, hiperinsülinizmin bir büyüme faktörü gibi rol oynayıp arteriel duvardaki proliferasyonu stimüle ederek makrovasküler hastalığı başlatabileceği, sodyumun renal tübüler reabsorbsiyonunu 13 hızlandırarak ve sempatik sinir sistemi aktivitesini arttırarak hipertansiyonun gelişmesine katkıda bulunabileceği ve son olarak karaciğerden çok düşük dansiteli lipoproteinlerin sentezini hızlandırıp ortamdan uzaklaştırılmalarını yavaşlatabileceği varsayılmaktadır. Arterioskleroz gelişmesi sonucunda koroner arter hastalığı ve serebrovasküler olaylar bu hastalarda sık olarak görülür. Ancak yukarıda sayılan durumların hepsi diabetes mellituslu hastaların hepsinde sık olarak görülen komplikasyonlar nedeni ile sendrom X’in kesin yöntemlerle ayırıcı tanısının yapılabilmesi bu gün için mümkün değildir. Tablo 5: Tip 2 diabet’in Etyolojik sınıflaması A) İnsülin Etkisine Göre 1- Glikoz klirensinde intraselüler defekt 2- İnsülin reseptör fonksiyonunda bozukluk a- İnsülin reseptör antikoru b- İnsülin reseptör mutasyonu (kromozom 19 p) 3- İnsülin yapısında bozukluk a- İnsülin gen mutasyonu (kromozom 11 p) insülin yapı anormalliği b- Proinsülinin insüline dönüşümünde bozukluk 4- İyatrojenik a- Glukokortikoidler b- Büyüme hormonu c- Nikonitik asit d- Diğerleri B) İnsülin Sekresyonuna Göre 1- Sinyal defekti a- Glukokinaz (hexokinaz IV) mutasyonu (kromozom 7 p) hücre kitlesinin yıkımı a- Otoimmun hücre yıkımı b- Pankreatitis (fibrokalkülöz pankreatik DM) c- Diğer sebepler C) Bilinmeyen Patogenesis 1- Malnutrisyon DM 2- Kistikfibrosiz 14 3- Talasemi 4- Hemokromatosis D) Tasnif Dışı (3) 1- İnsülin sekresyon ve etkisinde bilinmeyen nedenle azalma DİABETES MELLİTUS FİZYOPATOLOJİSİ(2) Diabetes mellitusun oluşumda birinci sebebin insulin yokluğu, yetersizliği veya insulin reseptörleri direnci olduğu bilinmektedir. Bu olayın etyolojik nedeni henüz kesin katları ile aydınlanmamıştır. Tip I diabette (insuline bağımlı) ve tip 2 diabette etyolojik neden ne olursa olsun sonuçta hiperglisemik tablo her iki tip diabetin en belirgin sonucudur. Hipergliseminin ve onun sonunda ortaya çıkan bozukluklar diabet oluşumunda karbonhidrat metabolizmasının büyük rolünü gösterir. Diğer yandan diabetik tablolarda, kan yağlarının ve proteinlerin katabolik gelişimi ve nihayetinde yağların yıkımından oluşan, keton cisimlerin hızla metabolize edilememesi ile ketoasidoz oluşumu, protein ve yağ metabolizmasının da etkilendiğini gösterir. Sonuçta diabetes mellitus bütün sistemleri ilgilendiren metabolik bir hastalıktır tanımını yapmak yanlış olmayacaktır. DİABETES MELLİTUSUN NEDENLERİ Diabetes Mellitus tip I (insuline bağımlı) ve tip 2 (insuline bağımlı olmayan) şeklinde iki gruba ayrılır. Bu grupların birbirlerinden ayrılmasında çok çeşitli faktörler rol alır. Başlangıç tabloları, heredite ile ilgileri HLA gen yapıları, çevresel faktörler, yaş ve klinik gelişimleri bu ayırımın yapılmasında önemli olup ilgili bölümlerde anlatılmıştır. Tip I diabette en önemli özellik Beta hücre yıkımıdır. Bunun sonunda insulin azlığı veya yokluğunun yanında insulin reseptörlerinde, insuline karşı normal olmayan cevaplar göze çarpar. 15 Tip 2 de en önemli özellik, insulin aktivasyonuna karşı hücrelerde direnç oluşumudur. Bu direnç insülin reseptörleri veya post insülin reseptör defektlerine bağlı olabilir. Tip 2 diabetin bir diğer özelliği de genellikle hücrelerinde insülin salınımında ve insülin yapısında bir bozukluk olmamasıdır. Bu tipte insülin azalması veya yokluğu hastalığın ileri devrelerinde immun çevresel faktörlerle pankreas dokularının azalmasına bağlanabilir. Diabetes mellitus tiplerinin yukarıda anlatılan özellikleri kesin sınırlarla ayrılmış değildir. Bazı olgularda insülin direnci gelişmiş tip I diabet ve insülin salgı azalması veya yokluğu gösteren tip 2 diabetes mellitus görülmesi de mümkündür. Tip I diabetes mellituslu hastalarda adacık topluluğunda selektiF olarak hücresi yıkımı ve insülin azlığı vardır. Bu hastalarda adacık hücrelerinden glUkogon salgılayan A ve somatostatin salgılayan D hücreleri korunmuş durumdadır. hücre yıkımının iki sebebi olduğu düşünülebilir. 1- Virüsler, 2- İmmün kompleksler. Çeşitli viruslar hücresi yıkımından sorumlu tutulmuşlardır. hayvan deneylerinde de diabetik hale getirilmiş farelerde etyolojik neden olarak kullanılan çeşitli virusler, hayvanların pankreas ve adacık dokularında tespit edilmiştir. Diğer yandan tip I diabetli hastaların, hastalıklarının her döneminde hatta balayı devresinde dahi kanda çeşitli antikorların tespiti, immun kompleksin etyolojik diğer bir neden olduğunu gösterir. Bu antikorlar hücrelerine karşı selektif etkili olduğu gibi, hücrelerine karşı sitotoksik etkiye sahip olabilir. Antijen, antikor kompleksinin oluşunun en önemli nedenlerinden biri virus enfeksiyonları sonucu olabilmesidir. KAN GLİKOZ DÜZEYİNİN KORUNMASI 16 Normal bir insanda 8-12 saatlik bir açlık devresinden sonra kan plazma glikoz değeri 70-120 mg/dl arasında ölçülür. Açlık değerleri daha uzun sürelerde bile bu denge korunur veya çok az miktarlarda glikoz seviyesi düşebilir. Bu denge gıda alımları sonrası da korunmakta olup, normalde gıda alımından sonra kan plazma glikoz seviyesi en fazla 160-170 mg/dl kadar yükselebilir. Kanın açlık ve tokluk devresi içinde glikoz seviyesindeki bu düzene glikoz homeostazı veya kanın normal glikoz düzeyi adı verilir. Kanın glikoz düzeyini sağlayan mekanizmalar ortaklaşa bir düzen içinde çalışırlar. Bu mekanizmalar: A. Kana glikoz sağlanması B. Kan glikozunun kullanılması C. Bazı hallerde kan glikozunun böbrekler yolu ile atılması NORMAL VE DİABETLİ İNSANLARDA METABOLİK DÜZENLEMELER Metabolik düzenlemeden kasıt maddelerin, temini depolanması, salınması ve end organlardaki kullanımı esnasında oluşan metabolik hareketleri izah etmektir. Karbonhidratların, proteinlerin ve yağların imali, depolanması ve gereken organlara sevkini sağlayan en önemli organ karaciğerdir. İnce barsaklardan portal ven yolu ile gelen ham materyal yani karbonhidratlar, proteinler ve yağlar karaciğerde değerlendirilerek ihtiyaca göre depolanma, birbirine dönüşme veya yakılmak üzere end organlara gönderilir. Glikoz yağlar ve proteinlerin birbirine dönüştürme, depolama ve yıkılma olaylarının, barsak, karaciğer ve end organlar arasında meydana gelmesiyle birçok hormonun önemli rolleri vardır. Bu olaylarda en etkili hormonlar, insülin, glukagon, büyüme hormonu, adreno kartikotropik hormon (ACTH), kortizon ve katekolamin hormonlar (Adrenalin, noradrenalin)'dir. İnsülin ön sıradaki etkisi kan şekeri düzeyini düşürmek için glikozun hücre içine girişini, karaciğerde glikojeni artırmasıdır. Bunun karşısında, glukogon, ACTH, büyüme hormonu, kortizon ve katekolaminler kan şekerini yükseltici etkiyi sağlarlar. Bu hormonlar vücutta kontrinsüliner sistemi oluştururlar. İnsülin ve kontrinsüliner hormonların etkileri 17 birbirleri ile olan oranlarına göre artar veya azalır. Kan glikozunun artışı insülin salınımını kamçılar, insülinin artışı insülin ile korteks hormonları ve büyüme hormonu arasındaki oranı büyütür. Bu halde glikojenoz hızlanır, glikozun dokulardaki kullanımı artar ve kan glikozu düşer. Aksi durumda kan şekerinin düşmesi, insülin salgılanmasını baskılar bu durumda insülin ile korteks, hipofiz hormonları arasındaki oran düşer. Glikojenez durur glikojenoliz artar. Dokularda glikoz yıkılımı en aza iner, sonuçta kan glikozu yükselir. KARACİĞER DOKUSU Glikoz NORMAL Glikoz-6-fosfat Glikoz Glikojen Trigliseridl er Trigliseridl er Glikoz DİABETLİ Glikoz-6-fosfat Glikoz Glikojen Trigliseridl er Trigliseridl er Yağ asidleri SYA Yağ asidleri NADH Piruvat asit SYA NADH Keton cisimcikleri Asetoasetat Piruvat asit Keton cisimcikleri Asetoasetat Asetil KoA Kolesterol Asetil KoA Kolesterol 18 A.A Üre Aminoasidl er A.A Aminoasidl er Üre Şekil 1: Karaciğer dokusunda normal ve diabetli ortamdaki metabolik değişiklikler Bu normal hormonal dengenin dışında acil mekanizmalar da görev alır. Kan şekerinin hızlı ve aşırı düşmesi durumunda oluşan hipoglisemide adrenalin ve noradrenalin ile glukogon hızla salgılanarak karaciğer glikojenolizini artırarak hayatı organların glikoz almasını sağlamaya çalışır. Beyin, eritrosit ve böbrek hücrelerinin glikoz kullanımı insüline bağlı değilken sadece kemik, kas ve yağ hücreleri glikoz kullanımı için kesinlikle insüline ihtiyaç duyar. Beyin böbrek ve eritrositler glikozu CO2 kadar yakarken, adele ve yağ dokusunda bu olay CO2’in anaerobik yoldan laksik aside kadar yıkılmasıyla oluşur.. Laktik asit, karaciğerde tekrar glikoza çevrilir. Karaciğer transaminasyon yolu ile yeni aminoasid, albumin yapımını da üstlenir. İnce barsak emilen karbonhidrat ve yağların aksine, emilen yağların ancak % 15'i vena portadan karaciğere gelir. Yağların % 85'i duktus torosikustan sol jugular ve subclavian venaya dökülür. Karaciğere gelen yağlar gliserol, serbest yağ asitleridir. Burada serbest yağ asitleri ve gliserol glikoza, gereğinde glikoz trigliseridlere dönüşür. Karaciğer serbest yağ asitlerini asetil koenzim A'ye dönüştürür. Asetil Koenzim A, keton cisimlerinin ( hidroksi bütirik asit, Asetoasetik asit aseton) sentezinde kullanılır. Ketoz cisimler kas dokusunda yakıt olarak yakılarak enerji sağlanır. Açlık halinde keton cisimleri beyin hücreleri içinde iyi bir yakıt kaynağıdır. Tüketim organlarından adaleler karaciğere alanin siklusu için alanin, böbrek, beyine glikoz, yağ dokusu da adaleye yakması için serbest yağ asitleri sağlayarak metabolizmayı dengede tutarlar. 19 NORMAL Glikoz DİABET Glikoz G1-6-F Dehidroksi Aseton Glikoz NADH2 Glikoz G1-6-F Dehidroksi Asetil-KoA Aseton fosfat NADH2 Asetil-KoA fosfat Trigliserid Trigliserid Yağ asitleri Yağ asitleri S.Y.A. Glikoz Alfa-glisero S.Y.A. Glikoz Gliserol Fosfat Alfa-glisero Gliseridler Gliserol Fosfat Gliseridler Şekil 2: Yağ dokusunda normal ve diabetli ortamdaki metabolik olaylar İNSÜLİN EKSİKLİĞİ Protein yıkımının artması Aminoasidemi ↓ Glikoneogenez artması Azalan glikoz utilizasyonu lipid sentezinin azalması ↓ ↓ Karaciğerde ve kaslarda glikojenoliz Lipolizin artması Hiperglisemi ↓ Glikozüri, osmotik diürez → ↓ ↓ Azot itrahını nartması Su ve elektrolit kaybı Fenallık kusma → Hiperglisemi Metabolik asidoz İnsasellüler dehitratasyon Dehidratasyon Hücrelerden K kaybı Hipovolemi ↓ 20 ↓ Hiperventilasyon Laktasitoz Kenonemi ↓ Ketonüri Na kaybı Dokularda hipoksi Periferik dolaşım bozukluğu Dokularda hipoksi Mutlak K eksikliği Debi renalin azalması Sürrenallerin stimulasyonu ↓ Beyin dolaşım bozukluğu Anüri ↓ İnsüline duyarlılığın azalması Koma ve ölüm Şekil 3: İnsülin eksiklğinin fizyopatolojik mekanizmaları TİP 2 DİABETES MELLİTUS PATOGENEZİ(3) Heterojen bir hastalık olan insüline bağımlı olmayan diabetin patogenezinden beta hücre fonksiyon bozukluğu, insülin direnci ve hepatik glukoz üretimi artışı gibi üç ana metabolik bozukluk sorumludur. Hepatik glukoz üretimi artışının primer defekt olduğunu gösteren bulgular azdır. İnsülin eksikliği ve/veya insülin direnci ise asıl nedeni oluşturur. Fakat NIDDM’un ortaya çıkışında insülin eksikliği ile seyreden beta hücre fonksiyon bozukluğundan veya insülin direncinden hangisinin primer olarak sorumlu olduğu güncel bir tartışma konusudur. Bunun yanında beta hücre fonksiyon bozukluğu ve insülin direnci arasında karşılıklı bir etkileşimin olduğu ve her ikisinin de patogenezde birlikte rol aldığı da ileri sürülmektedir. NIDDM’taki primer patolojinin beta hücre fonksiyon bozukluğu veya insülin direnci olmasında yaşın, etnik farklılıkların, şişmanlığın ve diabetin hetorejenitesinin kısmen de olsa belirleyici olduğu ileri sürülmektedir. Yukarıda belirtilen tartışmalardan ayrı olarak son yıllarda NIDDM’in oluşmasında dördüncü bir görüş olarak primer defektin hiperinsülinemi olduğu ve insülin direncinin hiperinsülinemiye bağlı olarak oluştuğu hipotezi ortaya atılmıştır. Bu hipoteze göre merkezi sinir sisteminde ventromedial hipotalamus, median eminence ve henüz tanımlanmayan bazı alanlardaki değişiklikler gıda alımı, termogenez ve 21 sempatik sinir sistem aktivitesinin düzenlenmesinde rol alan neuropeptid Y ve/veya diğer nöroregulatuar peptidlerin üretimini artırarak vagus sinirini uyarmakta ve bu da insülin salgısını uyarmaktadır. Ayrıca normal sağlıklı bireylerde yapılan çalışmalarda kronik fizyolojik öglisemik hiperinsülineminin insülin direncine neden olduğu gösterilmiştir. Hiperinsülineminin nonoksidatif glukoz kullanımını veya glikojen sentezini bozarak tıpkı NIDDM’de olduğu gibi insülin direncine yolaçabileceği ileri sürülmektedir. Fakat tüm bunlara karşın NIDDM’in oluşmasında en önemli iki patogenetik faktör insülin eksikliği ve insülin direncidir. 1. BETA HÜCRE FONKSİYON BOZUKLUĞU Normal glukoz toleransından bozulmuş glukoz toleransına ve hafif NIDDM’e geçildiğinde hiperinsülinemi oluşur. Açlık glukoz düzeyi 80 mg/dl’den 140 mg/dl’e yükseldiğinde insülin düzeyi normal sağlıklı bireylere göre 2-2.5 kat artar.Açlık glukoz düzeyi 140 mg/dl’i geçtiğinde ise beta hücreleri insülin salgılanması daha fazla artamaz ve açlık hiperglisemisi artıkça insülin salgılanması da kademeli olarak azalmaya başlar.İnsülin salgısının azalmaya başladığı bu sırada hepatik glukoz üretimi artmaya başlayarak açlık glisemisinin yükselmesine büyük katkıda bulunur. 250-300 mg/dl arasındaki açlık glisemi düzeyinde ise insülin salgılanması ciddi olarak azalır.İnsülin salgısındaki bu değişime Starling eğrisi adı verilmektedir. İnsülin direnci ile birlikte olsun veya olmasın eğer mutlak bir insülin eksikliği varsa NIDDM kaçınılmazdır. İnsülin salgılanmasında bozukluğa yol açan etyolojik faktörler aşağıda sıralanmıştır: İnsülin salgısında kantitatif bozukluklar İnsülin salgısında kalitatif bozukluklar Birinci faz insülin salgısının bozulması 22 Pulzatil insülin salgılanmasının bozulması Proinsülin salgılanmasında anomaliler Düşük doğum ağırlığı (Thrifty fenotip hipotezi) Glukoz toksisitesi Amilin (Adacık amiloid polipeptid) Calcitonin-Gene-Related–Peptid (CGRP ) İnkretinler (Glucagon like peptid-1, GİP, Galanin ) Lipotoksisite İnsülin salgılanma bozukluğunda genetik nedenler 1.İnsülin salgısında kantitatif bozukluklar NIDDM’un preklinik döneminde var olan insülin direncinin normale göre daha fazla insülin salınarak aşılmaya çalışılmasıyla normal glikoz toleransı sürdürülür. Bu dönemde öglisemik hiperinsülinemik klamp tekniği ile yapılan çalışmalar periferik insülin direncinin varlığını kanıtlamaktadır. Açlık glukoz düzeyi 80 mg/dl’den 140 mg/dl’e yükseldiğinde artan insülin düzeyi 140 mg/dl’den sonra hiperglisemiye bağlı olarak gittikçe azalır. 2. İnsülin salgısında kalitatif bozukluklar NIDDM’lu hastalarda insülin salgısının azalması yanında hedef dokuda insülinin etkisini potansiyalize eden insülinin salgı kinetiğinde de belirgin değişiklikler oluşur. Bunlar birinci faz insülin salgısının kaybolması ve pulzatil insülin salgılanması bozukluklarıdır. Birinci faz insülin salgısının bozulması İntravenöz glukoz verilmesini izleyen ilk 10 dakikada insülin salgılanmasında hızlı bir artış olur. İlk 2-4 dakikalar arasında zirve yapan insülin salgılanması 6.dakikadan sonra bu hızını kaybeder. Birinci faz insülin salgılanması adı verilen bu 10 dakikalık dönemden sonra insülin salgısı giderek azalmakta olup bu 23 süreçe de ikinci faz insülin salgılanması adı verilir. Birinci faz insülin salgılanması insülinin hedef bölgelerdeki etkisini potansiyalize eder.NIDDM gelişecek olanlarda birinci faz insülin salgısının kaybolması erken saptanabilen bir bulgudur. Bu defekt açlık plazma glukozu 115-120 mg/dl’i geçmedikçe oluşmaz. Burada ayrıca gecikmiş ikinci faz da mevcuttur. Birinci faz insülin salgısının kaybolması ile glukagonun hepatik glukoneogenezi arttırıcı etkisi belirginleşir. İkinci faz insülin salgılanmasının azalması ile de hepatik glukoz üretimi üzerindeki baskılayıcı etki azalır. Fakat 1.faz insülin salgı defektinin insülin direncinin patogenezinde de rol oynadığı ileri sürülmüştür. Yapılan bir çok çalışmada IGT’den NIDDM’e geçildiğinde insülin sensitivitesinde azalma ile birlikte 1. Fazı da içeren insülin salgılanmasında karşıt olarak artış görülmüştür. Ayrıca sıkı metabolik kontrolün 1. Faz insülin salgısını düzeltmesi bu defektin glukoz toksisitesi sonucu olduğunu düşündürmektedir . Pulzatil insülin salgılanmasının bozulması Normalde insülin her 5-15 dakikada bir periyodik olarak salgılanır. Salgılanma hızlı ve kısa süreli dalgalanmalar şeklinde olup glukagon düzeyi ile senkronizedir. Bu pulzatil salgılanma biçimi hedef dokularda insülin reseptörlerinin down-regulasyonunu önleyerek insülin sensitivitesinin normal sınırlarda kalmasını sağlar. Pulzatil olmayan sürekli insülin salgılanması ise reseptörlerde down-regulasyona yol açarak insülin direncine yol açar. NIDDM veya IGT’li bireylerde ve NIDDM’lu bireylerin birinci derecede yakınlarında bu hızlı ve kısa süreli dalgalanmalar yerine düzensiz ve daha kısa süreli dalgalanmaların oluşması karakteristiktir. NIDDM’lu obez hastalarda insülinin pulzatil salgılanmasındaki defektler kilo verilmesi ve sıkı metabolik kontrol ile büyük bir oranda düzelmekle birlikte tamamen normalleşmemektedir. 3. Proinsülin salgılanmasında anomaliler 24 Proinsülin insülinin ancak %5’i kadar biyolojik etkiye sahip olup insülin immünoreaktivitesinin normal bireylerde %2-4’ünü, NIDDM’lu bireylerde ise %8-10’unu oluşturur. Proinsülinin %70’ini 32-33 split (kırılmış) proinsülin oluşturur Proinsülin ve split proinsülinlerin klirensleri yavaş olduğundan ve de insülin ölçümünde kullanılan rutin RIA yöntemleri insülinin yanında proinsülinleri de (sağlam ve kırılmış) ölçtüğünden insülin düzeyleri olduğundan yüksek bulunur. NIDDM’de açlık total immünoreaktif insülin artışı ortaya çıkar bu da normal insülin düzeyleri üzerine eklenmiş olan artmış proinsülin düzeyinin bir sonucu olarak hiperinsülinemiyi gösterir. Gerçekte bu hiperinsülinemi olmayıp artmış proinsülin/insülin oranı göz önüne alındığında bir insülinopenidir. Proinsülinin parçalanmasında PC 3 (tip 1) endopeptidaz, PC 2 (tip 2) endopeptidaz ve karboksipeptidaz H olmak üzere 3 enzim rol oynamaktadır. PC 3 ve karboksipeptidaz H proinsülini 32-33 kırılmış proinsüline ve daha sonra da insülin ve C-peptide dönüştürmektedir. Hiperglisemi proinsülin ve PC 3 sentezi için güçlü bir uyarandır. PC 2 ve karboksipeptidaz H’ın sentezi ise glukoz tarafından düzenlenmemektedir. İnsülin direnci ve kronik hiperglisemi sonucu beta hücrelerinin sürekli uyarılması PC 3 aracılığı ile olan proinsülin sentezini artırarak 32-33 kırılmış proinsülin/insülin oranının artmasına yol açar. Dolayısı ile sürekli artan glukoz konsantrasyonlarında daha fazla proinsülin sentezlenecek ve proinsülini 32-33 kırılmış proinsüline dönüştürme yeteneğinde kompansatuar bir artış olmasına rağmen insüline dönüştürmede bir artış olmayacaktır. Buradan yola çıkarak plazmadaki sağlam ve 32-33 kırılmış proinsülin konsantrasyonlarının ölçümünün (çift işaretli immünometrik yöntemler ile) insülin direncine veya beta hücre salgılama kapasitesine ya da her ikisine bağlı olarak beta hücresinde oluşan fonksiyon bozukluğunu yansıtabileceği ileri sürülmektedir. Nitekim son yapılan bir çalışmada açlık 32-33 kırılmış proinsülin konsantrasyonlarının NIDDM’i insülin veya proinsülinden daha güçlü olarak predikte ettiği gösterilmiştir. 4. Düşük doğum ağırlığı (Thrifty-idareli fenotip hipotezi) 25 Thrifty (idareli) fenotip hipotezi sadece beta hücre yetmezliği olan bireylerin tip 2 diabete yakalanacağını öne süren bir hipotez olmakla birlikte son yıllarda yapılan çalışmalar düşük doğum ağırlığı ile erişkin yaşta ortaya çıkan IGT ve NIDDM arasında böyle bir bağıntının olabileceğini göstermektedir. Bu hipoteze göre fetüs ve bebeğin gelişimindeki yetersizliğin fetüs ve bebeğin yeterince beslenmemesine bunun da annenin yetersiz beslenmesine bağlı olduğu düşünülmektedir. Bu şekilde inutero malnütrisyona maruz kalan fetüs aldığı nütrisyonu idareli kullanmak için birtakım stratejiler geliştirerek beyin gibi hayati organlara öncelik vererek karaciğer ve pankreas gibi daha az hayati organların daha az beslenmesine yol açar. Sonuçta pankreas ve beta hücrelerinin yetersiz gelişimi düşük doğum ağırlığı ile sonlanır. Fötal gelişim sırasında sağlanan bu adaptasyon proğramı erişkin yaşamda beta hücresi için ek risk faktörlerinin eklenmesi ile bozulabilmektedir. Fetal nütrisyonun bozulması sonucu oluşan insülin sekresyonunda veya beta hücre kitlesinde azalmanın üzerine kalıtımsal olarak belirlenmiş olan insülin direncinin eklenmesi ile erişkin yaşta NIDDM’un oluşabileceği düşünülmektedir. Böylece normal yaşlanma süreci, obezitenin başlaması veya insülin direncinin genetik kompenentlerinin kötüleşmesi gibi faktörler ile beta hücresi insülin salgısını artırmaya başladığında eğer beta hücre kitlesi fetal hayatta gördüğü zarardan dolayı azalmışsa insülin salgısındaki artış yeterince olamıyacağından relatif bir hipoinsülinemi oluşur ve IGT veya Tip 2 diabet süreci başlar. İnsülin-signaling yolundaki proteinleri ve leptini kodlayan ve bu şekilde hipotezin genotipik temelini oluşturacak birkaç aday gen tanımlanmıştır. Bundan yola çıkarak thrifty genotipi ile konjenital lipoatrofik diabetin biribirlerinin ayna hayali olduğu (mirror image) düşünülmektedir. 5. Glukoz toksisitesi Hipergliseminin kendisi hem beta hücresi üzerine etki ederek insülin salgılanmasını baskılar hem de periferik dokularda insülinin kullanılmasını azaltır. Hipergliseminin beta hücresi üzerine olan bu olumsuz etkisine glukoz toksisitesi adı verilmektedir. Hiperglisemi durumunda sıkı metabolik kontrol ile (diyet, 26 sulfonilüre ve insülin tedavisi ile) insülin salgılanmasının düzeldiğinin gözlenmesi hipergliseminin kendisinin insülin salgılanması üzerine baskılayıcı bir etkisinin olabileceğini düşündürmüştür. Ayrıca yüksek glukoza sürekli maruz kalan beta hücresinde insülin gen transkripsiyonunun bozulduğu bunun da insülin sentezi ve sekresyonunu azalttığı gösterilmiştir. 6.Amilin (Adacık amiloid polipeptid) Amilin veya adacık amiloid polipeptid (IAPP) beta hücresindeki insülin salgı granüllerinde insülin ile birlikte üretilip beraberce salgılanan bir hormondur. Normalde bu hormon akut hiperglisemi sırasında veya diğer uyaranlara karşı insülin ile birlikte salgılanır. Amilinin kanda insülinden 1:10-50 gibi çok daha düşük bir seviyede bulunmasına rağmen insülinin etkisine karşı etkide bulunabileceği ya da insülinin etkisini inhibe edebileceği düşünülmektedir. Plazma amilin düzeyi obez glukoz intoleransı olan bireylerde, NIDDM’lu hastaların birinci derece glukoz intoleransı olan yakınlarında ve diyabetik hayvan modellerinde yüksek olarak bulunmuştur. Adacık amiloid depositleri tip 2 diabetik hastaların büyük çoğunluğunda gösterilmiştir. Bu amiloid depositlerinden en önemlisi adacık amiloid polipeptid (IAPP) olup ilk kez 1987 yılında tanımlanmıştır. Amilinin calcitonin gene-related peptid ile moleküler olarak % 46 benzerliği bulunmaktadır. Adacık amiloidinin apo E ve heparan sulfat proteoglycan perlecan olmak üzere iki önemli kısmı vardır. Amilinin (IAPP) hücre dışında beta hücrelerine bitişik olarak birikmeye başlayarak nütrientlerin plazmadan beta hücresine girişini engellediği ve sonuçta beta hücresinin ölümüne yol açtığı ileri sürülmektedir. Son yapılan bir çalışmada biriken bu IAPP agregatlarından özellikle küçük moleküllü IAPP depositlerinin olgun büyük moleküllü IAPP depositlere göre daha fazla sitotoksik oldukları gösterilmiştir. Human-IAPP solusyonu verilen sağlam adacıklarda da 24-48 saat içersinde apoptozis ve nekroz ile hücre ölümü olmaktadır. 27 Hipergliseminin kendisi de IAPP biyosentezini uyararak adacıklarda amiloidin daha da artmasına yol açar ve bu şekilde bir kısır döngüye sebep olur. Ayrıca hiperglisemik ortamda IAPP nin glycation’unun da amiloid fibril oluşumunu artırdığı ileri sürülmektedir. Transgenic mice‘larda yapılan ilginç bir çalışmada beslenmede artmış yağ alımının adacık beta hücrelerinde amiloid birikimine neden olabileceği bunun da insülinin sentez ve salgılanmasını bozarak diabete yol açacağı hipotezi ortaya atılmıştır. Ayrıca diyetteki yağın apoE biyosentezini artırarak amiloid fibrillerinin parçalanmasını azalttığı da gösterilmiştir. Fakat tip 2 diabetin patogenezinde amiloid birikiminin erken veya geç oluştuğu konusunda kesin bir kanıt bulunamamıştır. Tip 2 diabetli hastaların yaklaşık %100’ünde diabet olmayan yaşlıların da %10-20’sinde pankreaslarında amiloid birikimine rastlanmaktadır. İmmüno histokimyasal ve insitu hibridizasyon teknikleri kullanılarak diyabetiklerin çoğunda ve nondiyabetik kontrolerin %40’ında pankreatik dokudaki amiloid materyallerinden IAPP mRNA gösterilmiştir. Amilin bu amiloid artıklarının en önemli bileşkelerinden biridir. Bu veriler kronik hiperglisemi varlığında beta hücreleri içindeki amilin konsantrasyonunun arttığını ve amiloid birikiminin oluşma olasılığını göstermektedir. Bu tür birikimlerin oluşumu, insülin ve amilin salgılanmasını inhibe edebilir ve böylece beta hücrelerinin fonksiyonlarını durduruncaya kadar sürebilecek bir negatif ‘’feed back’’ döngüsü devreye girebilir. IAPP geninde NIDDM ile ilgili herhangi bir mutasyon bildirilmemiştir. İnsülinin hipersekresyonuna paralel olarak artan IAPP insülin reseptör antikorlarına bağlı olarak oluşan ağır insülin direncinde yaygın adacık amiloidozuna neden olmaktadır. Sonuç olarak tüm bunlara karşın amilinin NIDDM’ta beta hücre defektinden primer sorumlu olması kesin olarak gösterilememiştir. 7. Calcitonin gene related peptid (CGRP ) Amilinin calcitonin gene-related peptid ile moleküler olarak %46 benzerliği bulunmakla birlikte ratlara intravenöz olarak verildiğinde insülin salgılanması üzerine herhangi bir etkisi görülmemiştir. 28 8. İnkretinler (GLP-1,GİP, Galanin) Oral glikoz verildiğinde insülin sekresyonun artmasına neden olan faktörlere “inkretin” denir. Bunlar Glucagon-like peptide 1 (GLP-1), kolesistokinin ve gastrik inhibitör polipeptid (GİP) dir. Glucagon-like peptide 1 (GLP-1) ince bağırsakta sentez edilen potent insülin salgılatıcısıdır. Alınan besin maddeleri ile uyarılarak beta hücresi üzerindeki spesifik reseptörüne bağlanır ve adenilat siklazı uyarır bu da protein kinaz A’yı uyararak insülin salgılanmasına yol açar. NIDDM’lu hastalarda GLP-1’in glukoinkretin etkisi azalmakla beraber GLP-1 düzeyinin normal veya artmış olarak bulunması GLP-1’e karşı beta hücre rezistansı olduğunu göstermektedir. Farmakolojik dozda GLP-1’in NIDDM’li hastalarda postprandial insülin salgısını artırarak glisemiyi normal düzeylere yaklaştırır. Fakat GLP-1’in NIDDM’taki azalmış insülin salgısının patogenezindeki rolü için daha fazla araştırmalara gereksinim vardır. Güçlü bir glukoza bağlı insülin salgılatıcısı olan gastrik inhibitör polipeptid (GİP) farmakolojik dozda verildiğinde postprandiyal insülin salgılanması üzerine herhangi bir etkisi görülmemiştir. NIDDM’ta insülin salgısının bozulmasından en yeni olarak suçlanan hormon galanindir. Nöral uyarılara bağlı olarak pankreastaki sempatik sinir uçlarından salgılanan galaninin hayvanlarda bazal ve öğün sonrası insülin salgısını inhibe ettiği gösterilmiştir. Domuz galaninin insalara verilmesinden sonra glukoz ile uyarılmış insülin salgısı üzerine herhangi bir etkisinin olmadığı gösterilmiştir. Yine yeni olarak sentezi yapılan insan galaninin hiperglisemik klemp esnasında glukozla uyarılmış insülin salgısı üzerine bir etkisi gözlenmemiştir. Bu sonuçlardan yola çıkarak galaninin beta hücre fonksiyonlarının bozulmasında herhangi bir rolünün olmadığı düşünülmektedir. 9. Lipotoksisite Son yıllarda IGT’den NIDDM’e geçişte beta hücre fonksiyonlarında ilerleyici azalmayı açıklamak için tıpkı glukotoksisite gibi lipotoksisite kavramı ortaya atılmıştır. 29 Lipid metabolizmasındaki değişikliklerin glukoz ile uyarılmış insülin salgılanması üzerine önemli rolleri vardır. Sitozolik uzun zincirli fatty acyl-CoA esterleri insülin salgılanmasında tetiği çekici sinyal görevini üstlenirler. Serbest yağ asidleri (sFFA ), fatty acid binding protein 2 aracılığı ile beta hücrelerine taşınarak sitozolde fatty acyl-CoA ürünlerine çevrilirler. Daha sonra fatty acyl-CoA molekülleri carnitine palmitoyl transferase 1 (CPT-1) yardımı ile mitokondriye taşınarak krebs siklusuna girerler ve burada beta oksidasyona maruz kalırlar. Kan şekeri yükseldiğinde bu süreç inhibe edilerek sitozolik uzun zincirli fatty acyl-CoA konsantrasyonu yükselir ve bu da insülin salgısını uyarır. Artmış glukoz metabolizmasının bir sonucu olarak oluşan malonyl-CoA, carnitine palmitoyl transferase 1 (CPT-1)’i inhibe ederek sitozolik uzun zincirli fatty acyl-CoA’yı artırır. Uzun zincirli fatty acyl-CoA ürünleri spesifik protein kinaz C ‘yi direkt olarak aktive eden fosfatidik asid ve diacylglycerol oluşmasını artırır. Protein kinaz C de insülinin ekzositozunu artırır. Uzun zincirli fatty acyl-CoA esterleri K-ATPase kanallarının kapanmasını da uyararak insülin salgılanmasına yol açarlar. Yüksek düzeyde serbest yağ asidlerine maruz kalma sonucunda beta hücresinde trigliserid birikerek apoptozise yol açmaktadır. Yakın zamanda yağ asidlerinin, proinsulinin insüline çevrilmesinde (ve proIAPP’nin IAPP ye çevrilmesinde) rol alan PC1/3 ve PC2 endoproteazlarının posttranslational işlemini azalttıkları bildirilmiştir. Sonuç olarak glukoz ve FFA insülin salgılanmasını artırmakta fakat bir süre sonra uzun zincirli fatty acyl-CoA’yı artırarak down-regulasyona yol açmakta ve/veya Randle siklusu yolu ile insülin salgılanmasını inhibe etmektedir. Bu şekilde beta hücrelerinin artmış FFA düzeylerine uzun süre maruz kalması olarak adlandırılan lipotoksisite insülin salgı bozukluğunun önemli sebepleri arasında gösterilmektedir. 10. İnsülin salgılanma bozukluğunda genetik nedenler Glukozun beta hücresi tarafından tanınmasında, insülinin sentez ve salgılanmasında rol oynayan spesifik proteinlerdeki mutasyonlar beta hücresi disfonksiyonundan sorumlu olabilmektedirler. Şimdiye dek glukokinaz geni, mitokondriyal DNA geni ,insülin geni ve insülin proçesindeki enzimlere ait genlerde 30 mutasyonlar tanımlanmıştır. Bu mutasyonlar oldukça nadir olup tüm NIDDM’lerin %1-2 ‘sini oluştururlar. Glukokinaz geninde çeşitli defektlerin gösterildiği üç ayrı MODY tipi tanımlanmıştır. Mitokondriyal DNA gen mutasyonunda miyopati, sağırlık ve nörolojik anomaliler olup maternal olarak geçer. Bunlarda insülin salgılanması bozulmakla beraber mekanizması tam olarak bilinmemektedir. Beta hücrelerinin glukozu tanımasını kolaylaştıran ve böylelikle insülin salgılanmasını sağlayan GLUT 2 proteinin de bir mutasyon gösterilememiştir. İnsülin ve glukokinaz genlerindeki mutasyonların sonucunda beta hücre fonksiyonunda oluşan kısmi bozukluk NIDDM gelişimine katkıda bulunabilir. İnsülin genindeki mutasyonlar biyolojik aktivitesi bozulmuş insülin sentezine yol açarken, glukokinaz genindeki mutasyonlar beta hücrelerinin glukozu tanıma fonksiyonunu bozmaktadır.Beta hücresindeki glukokinaz gen promoter değişikliklerinin insülin salgılanmasında bozukluğa yol açtığı gösterilmiştir. Bu genlerdeki mutasyonlar heterozigot durumlarında saptanmış olup bu durumda beta hücre fonksiyonları yarı yarıya korunur. Bu nedenle genlerinden herhangi birinde mutasyon olan bireyler %50 oranında hafif diabet riski taşırlar. Kan Şekeri Kontrolü Neden Bu Kadar Önemlidir? Açlık durumunda insülin üretiminin supresyonu ve glukagon üretiminin stimulasyonu, kan glukoz konsantrasyonunu kontrol eder. Bu süreçler karaciğerin glikojen depolarından glukoz mobilizasyonuna ve aminoasitler ve piruvattan glukoz sentezine (glukoneogenez) izin verir. Ayrıca insülin düzeyleri düşük olduğunda glukozun kaslar tarafından alınması en az düzeye iner ve adipozitler serbest yağ asitleri sağlarlar. Bu homeostatik mekanizma açlık durumunda beyine yeterli glukoz desteğini sağlamak amacıyla (beyinin enerji deposu yoktur) plazma glukoz seviyesini stabil olarak tutar. Beslenme durumunda insülin iki fazda salınır. Kısa, yiyecek alımı veya plazma glukoz konsantrasyonunda bir artış durumunda küçük patlamalar tarzındaki birinci faz postprandial glukoz 31 artışlarını azaltır. Daha sonra, daha uzun süren ikinci fazla insülin salınımı başlar. Bu bifazik insülin salınmına cevapta karaciğer glukozu alır ve glikojene çevirir. Kas ve yağ dokuları da glukozu alır ve sırayla glikojen ve trigliserit olarak depolar. Dahası adipozitlerde, serbest yağ asitlerinin üretimi baskılanır. İkinci faz insülin salınımı yeterli hatta artmış olsa bile birinci faz insülin salınımının kaybı metabolik ve fizyolojik sonuçlara yol açar. Eksojen İnsülin Tedavisi Tip 1(4-9) ve tip 2 diabetes mellitus(10, 11) hastalarında diabet tedavisinin amaçları; hastayı semptomsuz kılmak, yaşam kalitesini yüksek tutmak, iyi bir glisemik kontrolle komplikasyon riskini en aza indirmektir. Hastaların ve sağlık personelinin karşılaştığı sorunlardan biri, iyi bir glisemik kontrol sağlayabilmek amacıyla uygulanan yoğun insülin tedavisine bağlı artmış hipoglisemi riskidir(5). Varolan insan insülini formülasyonlarıyla yapılan eksojen insülin tedavisi normal fizyolojik cevabı yakından taklit edebilen serum insülin düzeyleri ortaya koyamamaktadır. İnsülin gereksinimi olan hastaların çoğu kısa etkili bir insülinle (regüler) bazal insülini (orta ya da uzun etkili) kombine kullanmaktadır. Kısa etkili insülin, genelde postprandiyal glukoz dalgalanmalarını karşılayabilecek serum insülin düzeyini oluşturabilmek için yemek öncesi kullanılır. Ancak; regüler insan insülininin subkutan enjeksiyonu; diabetik olmayan kişilerdeki normal pankreatik insülin salınımını takiben gelişen serum insülin konsantrasyonlarına göre daha geç zirve düzeye ulaşmakta (enjeksiyondan 3-4 saat sonra) ve daha uzun süre yüksek (etki süresi 7-8 saat) kalmaktadır(12-13). Sonuç olarak; postprandiyal dönemde saatlerce süren ve potansiyel hipoglisemi riskine yol açan göreceli hiperinsülinemi olabilir ve klinik pratikte bu durum nedeniyle hasta öğün aralarında atıştırmak zorunda kalabilir. Hipoglisemiye yol açma riskini en aza indirgemek için subkutan regüler insan insülin enjeksiyonlarının genellikle yemeklerden yarım saat önce yapılması önerilir(14). Özelikle esnek ve değişken bir yaşam biçimi olanlar olmak üzere tüm hastalar için bu uygulama tarzı uygun değildir. Çoğu hasta preprandiyal dozları, önerilenden daha erken kullanırlar(15,16) ve böylece regüler insan insülininin 32 ideal zaman-etki profiline uymadıklarından yan etki gelişme olasılığı artmış olur. İNSÜLİN LİSPRO İnsülin lispronun (Humolog Penfill, Lilly, USA, ındiana police) geliştirilmesindeki temel amaç; diabetik olmayan bireylerdeki fizyolojik postprandiyal insülin cevabına yakından benzerlik gösteren bir terapötik yaklaşım üretebilmektir. İnsülin lispro regüler insan insülinine göre etkisinin daha hızlı başlaması ve daha kısa sürmesi yoluyla; glisemik kontrolü iyileştirir, hipoglisemik atak sıklığını azaltır, hastanın tedavi memnuniyetini artırır ve hiperglisemiyi daha hızlı kontrol altına alır. Çözelti içindeki insan insülin molekülleri kendi kendilerine ayrışma eğilimindedirler. İki molekül birbirlerine çok yakın olduklarında, aralarında geri dönüşümlü bir bağ gelişerek insülin dimeri ortaya çıkmış olur. Piyasadaki insan (ve hayvan) insülini preparatlarında olduğu üzere çinkonun varlığında; üç dimer heksamer oluşturmak üzere ayrışırlar(17). Subkutan enjeksiyon sonrasında, insülin molekülleri sistemik dolaşıma katılmadan önce insülin heksamerleri ayrışmalıdır. İnsülin heksamerlerinin ayrışmaları için gereken zaman regüler insan insülini zaman- etki profilini yansıtmaktadır. Daha hızlı etkili bir insülin bulabilmek için yapılan incelemeler kendi kendine ayrışma eğilimi daha az olan bir insülin molekülü geliştirmek üzerinde odaklandı. İnsan insülininin B zincirindeki prolin ve lizin adlı aminoasitler göreceli olarak 28. ve 29. pozisyonda bulunmaktadırlar. Prolin ve lizinin yerini değiştirmek, insan insülinine göre kendi kendine ayrışma eğilimi daha düşük olan bir insülin analogunun, insülin lispronun gelişmesine yol açtı(18). Kendi kendine ayrışma eğilimi oldukça düşük olduğu halde insülin lispro çinko ve koruyucu madde varlığında, çözelti içinde heksamerler oluşturabilir. Daha da şaşırtıcı olarak, heksamer halindeki insülin lispro, subkutan enjeksiyon sonrasında heksamer halindeki insan insülininden daha hızlı monomerlerine parçalanır(17). Bu hızlı ayrışma ve emilim insülin lispro heksamerlerindeki fenolik koruyucuların insülin heksamerleriyle karşılaştırıldığında farklı rol oynamasıyla açıklanabilir(17). 33 Subkutan enjeksiyon sonrasında regüler insan insülinine göre daha hızlı emilir, etkisi daha çabuk başlar ve daha kısa süre etkili olur(19). Sonuç olarak; insülin lispro kısa etkili insülinlerden daha fizyolojik bir zaman, etki profiline sahiptir ve tedavinin optimizasyonunun sağlanmasında atılmış bir adım olarak değerlendirilebilir. İnsülin lispro, glukoz homeostazının sağlanması için insüline gereksinim duyan diabetes mellituslu hastaların tedavisinde ve diabetes mellitusun başlangıç stabilizasyonunda endikedir. İnsülin lispro uzun etkili bir insülinle birlikte de kullanılabilir ve preprandiyal kullanılması önerilir. Kimya ve Formülasyon İnsülin lispro (Lys (B28), Pro(B29))-insan insülini (rekombinan DNA kaynaklı), B-zincirinin Cterminalindeki prolin (B28) ve Lizinin (B29) yer değiştirmesi sonucu oluşan biyosentetik bir insan insülin analogudur. Bu aminoasitler insülin preparatlarının kendi kendine ayrışma davranışında önemli rol oynarlar ve bunlarla ilgili yapılan bir manipulasyon kendi kendine ayrışma eğiliminde azalmayla sanuçlanır. İnsan insülininde B28 pozisyonunda bulunan Prolin insülin dimerinde oluşan beta zincir düzeninin olusumunda ve stabilizasyonunda önemli rol oynar. Bu moleküller beta zincir yüzeyinde antiparalel alarak yerleşmişlerdir(20). Çinkonun varlığında; üç dimer ayrı heksamerler oluşturmak üzere parçalanır(20). İnsülinin heksamerik durumu bulunan tüm regüler insülin formüllerinin temelini teşkil eder(21). İnsülin lisproda yapılan dizisel yer değiştirme prolini B28’den B29 pozisyonuna getirir ve dimerin monomer-monomer yüzeyinde oluşacak iki kritik hidrofobik etkileşim önlenmiş olur(17). İnsülin lispronun B zincirinde aminoasit dizisinin değişmesi, insülin lispronun hidrofobik etkileşimini ortadan kaldırır ve moleküllerin kendi kendilerine ayrışma eğilimini büyük ölçüde azaltır. Beklenmedik bir şekilde; insülin lispro heksamerinin, analogun çinko ve fenolik koruyucularla formüle edilmesiyle de oluşabileceği görülmüştür. Böyle bir heksamer kartuşta ya da flakonda insülinin fiziksel ve kimyasal açıdan stabil olmasını sağlar. Stabil bir heksamer olmasına rağmen, insülin lispro, subkutan enjeksiyon 34 sonrasında regüler insan insülininden daha hızlı bir şekilde monomerlerine ayrışır. Böylece insülin lispro çok daha çabuk emilmiş olur(19). İnsülin lispronun farklılaşmış kendi kendine ayrışma özelliğine ait yapısal deliller, insülin lispro heksamerinin kristal yapısından elde edilebilir(17). Sentez İnsülin lispro, insülin lispro öncülü bir genin eklenmesiyle değişime uğramış Escherichia coli bakterilerinin hastalığa neden olmayan neslinden sentezlenir. İnsülin lispro öncülü konaktan izole edilir ve pek çok işlem sonrasında saflaştırılarak çinko-insülin lispro kristallerine dönüstürülür. Bu kristaller ilacın son haline gelebilmesi için formüle edilirler. Stabilite ve Depolama Insulin Iıspro buzdolabında 2 ila 8 C0 de saklanmalıdır. Aşırı sıcak ya da güneş ışığına maruz kalmamalı ya da dondurulmamalıdır. Buzdolabı yoksa, kullanılmış insülin lispro flakonu veya kartuşu oda ısısında (30C0’ nin altında ve direkt ısı ve ışıktan uzakta) 28 güne kadar saklanabilir. Enjeksiyon kalemine yerleştirdikten sonra kartuş ve kalem buzdolabına konulmamalıdır. İnsülin lispro donmuşsa ya da etiketinde yazılı olan son kullanma tarihi geçmişse kullanılmamalıdır. Klinik Farmakoloji İnsülin lispronun farmakokinetik ve glukodinamik profilleri; diabetik olmayan kişilerde ve tip 1 veya tip 2 diabeti olan hastalarda, regüler insan insülinine göre etkisinin daha çabuk başladığını, zirve aktiviteye daha çabuk ulaştığını ve etki süresinin daha kısa olduğunu göstermişlerdir. Regüler insan insülini ile karşılaştırıldığında, insülin lispronun etkisi, subkutan enjeksiyon yerinden bağımsız olarak daha cabuk baslar ve daha kısa surer. İnsülin lispro, regüler insan insülini ve domuz insülini ile gelişen hipoglisemiye karşı oluşan karşı düzenleyici cevaplar önemli düzeyde farklı değillerdir. İnsülin lispro veya regüler insan insülininin farmakokinetik profiliyle bağlantılı olarak neden olduğu hepatik ya da renal hasar minimaldir. Terminal evre böbrek hastalığında, hem insülin lisproya hem 35 de regüler insan insülinine yanıt uzar. Yapılan çalışmalarda subkutan enjeksiyon sonrasında insülin lispronun regüler insan insülininden daha hızlı emildiğini(19), etkisinin daha çabuk başladığını, zirve aktiviteye daha çabuk ulaştığını ve daha kısa süre etkili olduğunu göstermişlerdir. Zirve aktivite subkutan enjeksiyondan yaklaşık 1-3 saat sonra başlar ve yaklaşık 4-5 saat sürer. İnsülin lispro ile regüler insan insülini arasındaki farmakokinetik ve glukodinamik farklılıklar diabetik olmayan kişilerde ve diabetes mellituslu hastalarda belirgindir(19). İnsülin Lispronun NPH ve Uzun Etkili İnsülinlerle Kombinasyonu Glukoz klamp-kontrollü çalışmalar; insülin lispro ve NPH (orta- etkili insülin)(22) ya da Ultralente (uzun etkili insülin) kombinasyonlarının ya da ayrı ayrı subkutan enjeksiyonlarının farmakokinetiğini ve glukodinamiğini karşılaştırmak amacıyla düzenlenmiştir(23). lnsülin lispro ve orta etkili ya da uzun etkili insülin ya aynı anda farklı enjeksiyon yerlerinden ya da tek bir enjektörde karıştırılarak uygulandı. Her iki durumda da insülin lispro daha hızlı emildi. lnsülin lispronun NPH ya da Ultralente ile kombinasyonu ile ayrı ayrı verilmeleri arasında hiçbir farmakokinetik ve glukodinamik farklılık saptanmaması; insülin lispronun NPH veya Ultralente ile aynı enjektörde karıştırılması halinde farmakokinetik ve glukodinamik aktivitesini koruduğunu göstermektedir. İnsülin Lispro ve Eşlik Eden Tedaviler Oral Hipoglisemik Ajanlar Tip 2 diabetli ve sülfonilüre tedavisi başarısızlıkla sonuçlanan hastalarda yapılan bir randomize, açık ve çapraz çalışma; postprandiyal hiperglisemideki iyileşmenin günlük total glukoz kontrolünü etkileyip etkilemeyeceğini belirlemek amacıyla yürütüldü(24). Araştırmaya katılanların bir kolu sülfonilüre kullanmaya devam ederken, diğer kol insülin lispro ve sülfonilüre kullanmaya başladı. Hastalar dört hafta sonra tedavileri değiştirdiler. Sülfonilüre ile birlikte kullanılan insülin lispro, 2-saatlik postprandiyal glukozu tek başına sülfonilüre kullanımına göre önemli ölçüde düşürdü (18.6 dan 14.2 mmol/L’e, p<0.0001). HbAıc, insülin lispro-sülfonilüre kombinasyonu ile tek başına sülfonilüreye göre 36 daha fazla düştü (% 9.0’dan 7.1’e, p<0.0001). Randomize açık ve iki dönemli, 423 hastada yapılan bir çalışmada; oral ajanla tedavi başarısız olduktan sonra insülin tedavisine gereksinim duyan tip 2 diabet hastalarında üç tedavi rejiminden hangisinin en etkili olduğunu ortaya koymak amacıyla yürütüldü. Hastalar, ya yemek zamanında insülin lispro ile kombine gece NPH insülin veya günde iki kez sülfonilüre ya da gece NPH insülin ile kombine günde iki kez sülfonilüre kullandılar. Tedavi başarısı değerlendirildiğinde; insülin lispronun NPH insülin ile kombinasyonunun daha iyi cevap verdiği gözlendi. Ek olarak, hemoglobin A1c sonuçları, insülin lispronun NPH insülin veya sülfonilüre ile yaptığı kombinasyonlarla uzun dönem daha iyi glisemik kontrol sağladığını ortaya koydu. Ancak, bu değişikliklerin hiçbiri istatistiksel açıdan önemli değildi. Beş noktalı kan glukoz profilleri; her iki insülin lispro tedavi grubunda kısa dönem glisemik kontrolün daha iyi olduğunu gösterdi. Bu iki grupta kan glukoz düzeyleri yemek sonrasında önemli ölçüde yükselmedi. İki insülin lispro tedavi grubunun karşılaştırılması sonucunda da benzer etkililik profili elde edildi. Total olarak, yan etki veya ciddi yan etki açısından tedavi grupları arasında farklılık bulunamadı. İnsülin lisproyu NPH insülin ya da sülfonilüre ile kombine kullanan hastalar, NPH insülinini sülfonilüre ile birlikte kullanan hastalara göre az, ancak klinik anlamı olmayan bir kilo artışı gösterdiler. NPH insülinsülfonilüre kombinasyonu kullanan hastalarda hipoglisemik atak insidansı, insülin lispro kullananlardan daha azdı(25) Yüzotuzbir hastayla yürütülen diğer bir randomize, açık ve iki dönemli çalışma, oral ajanla tedavi başarısız olduktan sonra insülin tedavisine gereksinim duyan tip 2 diabet hastalarında üç tedavi rejiminden hangisinin en etkili olduğunu ortaya koymak amacıyla yürütüldü. Hastalar günde iki kez gliburid ya yemeklerde insülin lispro ya yatarken NPH insülin ya da günde iki kez metformin ile kombine olarak kullandılar. İnsülin lispro ile yapılan kombinasyon, NPH insülinle (p=0.003) veya metforminle (p=0.025) yapılan kombinasyonlarla karşılaştırıldığında, HbAıc’yi önemli ölçüde daha fazla düşürmüştü. NPH insülin ile yapılan kombinasyonla, son nokta açlık kan glukoz düzeyi; insülin lispro (p<0.001) ile 37 veya metformin (p=0.029) ile yapılan kombinasyonlarla karşılaştırıldığında, önemli ölçüde daha düşüktü. İnsülin lispro ile yapılan kombinasyonla, test yemeği sonrası ortalama 2 saatlik postprandiyal glukoz, NPH insülinle (p=0.052) veya metforminle (p=0.009) yapılan kombinasyonlara kıyasla önemli ölçüde daha düşüktü. İnsülin lispro veya NPH insülinle kombine tedavileri kullanan hastalarda, metformin kombinasyonu kullanan hastalara göre, küçük, ancak klinik önemi olmayan bir kilo artışı gözlendi. Total hipoglisemi hızı düşüktü ve gruplar arasında istatistik açıdan farklılık göstermedi(26). Diğer İlaçlar Tip 2 diabetes mellituslu hastaların yaklaşık % 15’i antihipertansifleri de içeren ek bir ilaç kullanmaktadırlar. Aşağıdaki bilgiler insülinlerle uyumlu olup insülin lispro için de geçerlidir: Hipoglisemi; diabetli bir hastanın insülin tedavisi sırasında en sık yaşayabileceği istenmeyen etkidir. Hipoglisemi bilinç kaybına ve ciddi vakalarda ölüme neden olabilir. Hastalarda lokal alerji, enjeksiyon yerinde kızarıklık, şişme ve kaşıntı olarak kendini gösterir. Bu durum, günler ya da haftalar içinde düzelir. Bazı durumlarda, bu alerji derinin temizlendiği maddeler içindeki irritanlara veya enjeksiyon tekniğinin kötü olması gibi insülin dışı etmenlere bağlı olabilir. Daha nadir ancak çok daha ciddi olma potansiyeli olan sistemik alerji ise insüiine karşı gelişen genel bir alerjidir. Tüm vücutta döküntü, nefes darlığı, hırıltı, kan basıncında düşme, nabızda hızlanma veya terlemeye neden olur. Ciddi vakalar yaşamı tehdit edici olabilir. Enjeksiyon yerinde lipodistrofi gelişebilir. Fazla doz: insülinlerin belirli fazla doz tanımları yoktur, çünkü serum glukoz düzeyi, serum insülin düzeyi, glukozun bulunabilirliği ve diğer metabolik süreçler arasındaki karmaşık bir etkileşim sonucunda belirlenir. Hipoglisemi, yenilen yemeğe ve harcanan enerjiye göre fazla insülin lispro kullanılması sonucu gelişmiş olabilir. Dozaj, Uygulama, Kontrendikasyonlar, Uyarılar ve Önlemler 38 İnsülin lispro preprondiyal kullanım için aşağıdaki durumlarda endikedir: Normal glukoz homeostazı için insül ine gereksinim duyan diabetes mellitus hastalarının tedavisinde Diabetes melhtusun başlangıç stabilizasyonunda insülin lispro hızlı etkili bir insülin analogudur ve uzun etkili bir insan insülini ile birlikte kullanılabilir. İnsülin lispronun dozu hastanın gereksinimlerine göre doktoru tarafından belirlenmelidir. İnsülin lispro subkutan yoldan kullanılmalıdır. Onerilmemesine karşın intramüsküler olarak ta kullanılabilir. İnsülin lispronun subkutan enjeksiyonu karın, üst kol, baldır veya kalçalara yapılmalıdır. Enjeksiyon yerleri belli aralarla değiştirilmelidir. Böylece aynı yer ayda birden daha sık kullanılmamış olur. İnsülin lispronun etkisi regüler insan insülininden daha önce başlar ve daha kısa sürer. Etkisinin erken başlaması nedeniyle insülin lispro yemek zamanına çok yakın zamanlarda kullanılabilir. Herhangi bir insülinin etkisinin başlaması farklı bireylerde ve hatta aynı bireyde farklı zamanlarda önemli farklılıklar gösterebilir İnsülin lispro uzun etkili bir insan insülini ile birlikte kullanılabilir. İnsülin lispro kullanımının kontrendikasyonları; hipoglisemi ve insülin lispro veya katkı maddelerine karşı aşırı duyarlılıktır. Bir hastanın başka tip bir insüline geçişi katı bir tıbbi denetim altında yapılmalıdır. İnsülinin gücünde markasında (üretici), tipinde (regüler, NPH vs.), türünde (hayvan, insan, insan insülin analogu) ve/veya üretim yönteminde ( rekombinan DNA, hayvan kaynaklı insülin) yapılan bir değişiklik insülin dozlarında değişiklik yapmaya neden olabilir. Ayarlama gerekiyorsa, ilk dozda ya da insülin lispro ile tedaviye başlandıktan sonraki haftalarda veya aylarda yapılır. 39 İnsülin lispro uzun etkili bir bazal insülinle karıştırılırsa, şişenin uzun etkili insülinle kontaminasyonunu önlemek için enjektöre önce insülin lispro çekilir. Kan glukozu çok düzelen hastalarda (yoğun insülin tedavisi ile) hipogliseminin bazı ya da tüm uyarıcı belirtileri ortadan kalkobilir, hastalar bu duruma karşı uyarılmalıdırlar. Hayvan kaynaklı insülinden insan kaynaklı insüiine geçtiğinde hipoglisemik atak geçiren az sayıda hasta, hipogliseminin erken uyarıcı semptomlarının daha az belirgin ya da ilk kullandıkları insülinle yaşadıklarından farklı olduğunu bildirdiler. Düzelmemiş hipoglisemi veya hiperglisemi ciddi tıbbi sorunlara neden olabilir. İnsülin gereksinimi renal veya hepatik bozukluk hallerinde düşebilir. İnsülin gereksinimi hastalıkta veya emosyonel bir bozuklukta artabilir. Hasta alışageldiği diyetini değiştiriyorsa, normal rutinin dışında bir fiziksel aktivitede bulunuyorsa doz ayarlaması gerekebilir. İnsülin gereksinimi; oral kontraseptifler, kortikosteroidler, tiroid replasman tedavisi, danazol, ve 6-2 stimulanları gibi hiperglisemik aktivitesi olan ajanlarla artabilir. İnsülin gereksinimi; oral hipoglisemikler, salisilatlar, sülfa antibiotikleri, bazı antidepresanlar, bazı anliotensin dönüştürücü enzim inhibitörleri, beta blokerler, oktreotid ve alkol gibi hipoglisemik aktivitesi olan ilaçların varlığında azalabilir. İnsülin lispro hayvan insülinleriyle karıştırılmamalıdır. Hamilelikte İnsülin Lispro Hamilelikte insülin lispro kullanımına dair ciddi bir deneyim bulunmamaktadır. Diabeti olan hamilelerde glukoz kontrolünün ve genel sağlığın dikkatle izlenmesi gereklidir. 40 MATERYAL VE METOD Çalışmaya Şişli Etfal Eğitim ve Araştırma Diabet polikliniğine 2003-2004 yılları arasında başvuran ve yeni tespit tip 2 diabet tanısı almış olan hastalar alındı. Çalışmamız, tek merkezli, randomize, açık, prospektif bir çalışma olarak planlandı. Hastalar iki gruba ayrılarak birinci gruba OAD, ikinci gruba ise yemek zamanı 3 kez hızlı etkili insülin analoğu ve yatarken bir kez NPH insülin uygulandı. HbA1c >%7 olan 10 hasta başvuru sırasına göre randomize edilerek ve hastalardan yazılı olur onayı alınarak (iyi klinik uygulamalarda ‘International Committee of Harmonitation Good Clinical Practice’ belirtilen esaslar doğrultusunda ) çalışmaya alındı ve altı hafta süreyle izlendi. Hastalara tedavi başlamadan önce ve 6 hafta sonunda; -AKŞ : end point kalorimetrik yöntem ile, -TKŞ(1.saat): end point kalorimetrik yöntem ile, -Bazal C-peptit düzeyleri. Radyoimmunassay yöntemi ile intraassay CV %3.1, interassay CV %5.2 (İmmunotech C-Peptide Kit, Marseille, Cedex, france), -Glukagonla uyarılmış c-peptid düzeyleri(6.dakika), -Proinsülin(0.dk ve 6. dk): İntrassay CV %4.3, Interassay CV %5.5, Low detectibl limit: 0.5 p/mol/L (Immuno Biological Laboratieries. Hamburg, Germany ), -HbA1C : İmmunotirbüdimetrik yöntem ile , 41 -Üre, kreatinin: Otoanalizörde kalorimetrik yöntem ile, -Kolesterol, trigliserit,LDL,HDL: end point kalorimetrik yöntem ile ölçüldü. -BMI’leri bakıldı. (kg/m2( vücut ağırlığının kg cinsinden değerinin, vücut yüzey alanının m2 cinsinden değerine bölünmesi ile elde edilir. Test Uygulamaları: -Glukagon testi; Testler için Glukagon Hypokit (Novo Nordisk, Copenhagen, Denmark) kullanıldı. Hastalara sabah 1 gece açlık sonrası (hastalara o gece ve sabah insülin yapılmadı) Proinsülin ve C-peptit için glukagon öncesi örnekler alındı. 1 mg IV glukagon sonrası 6. dakikada testler tekrar edildi. Hastalar: Yeni tanı konmuş toplam 20 tip 2 Diabetes Mellituslu hasta randomize edildi. 10 hasta insülin grubuna alınırken, 10 hasta OAD grubuna alındı. İnsülin grubuna 3 kez hızlı etkili insülin analoğu (Insülin Lispro (Humolog Penfill, Lilly, USA) ve geve NPH (Humulin N penfill, Lilly, USA) verildi. OAD grubuna glimeprid 2 mg 8Amaryl 2 mg, Aventis, France) başlandı. Alınma Kriterleri: 1.Yeni tanı almış Tip II Diabetes Mellitus 2.Diabetik komplikasyon oluşmamış 3.BMI ≤ 35 4.HbA1c ≥ % 7 5.Yaş < 60 6.C -peptid>3.5ng/ml Dışlama Kriterleri: 1.Herhangi bir düzeyde renal yetersizlik 2. Herhangi bir düzeyde hepatik yetersizlik 42 3.Kalp Yetersizliği 4.Alkolizm 5.Malignite 6.Kronik enfeksiyonlar 7.Astma Bronşiale Proinsülin düzeyi bakılırken, “IBL(Immuno-Biologiacl Laboratreies) Proinsulin ELISA” kiti kullanıldı. Proinsülin < 9.4 pmol/L ise normal olarak kabul edildi. C-peptid düzeyi ise “Immunotech C-peptide” kiti ile radioimmunassay yöntemi ile belirlendi. Cpeptid bazal düzeyinin bazal değerleri 0,48-3,80 ng/mL arasında normal olarak değerlendirildi. HbA1c düzeyi mikrokolon yöntemi ile bakılarak %4.00-6.00 değerleri normal sınırlarda kabul edildi. Diğer biyokimyasal parametreler biyokimya otoanalizatöründe bakılarak referans aralıkları ve birimleri aşağıdaki tablodaki gibi değerlendirildi.. Tablo 6. Biyokimyasal parametrelerin normal değerleri TEST GLUKOZ / AÇLIK Birim mg/dL Minimum 75.0 Maksimum 110.0 GLUKOZ / TOKLUK mg/dL 75.0 140.0 BUN mg/dL 7.0 18.0 ÜRE mg/dL 15.0 45.0 KREATİNİN mg/dL Erkek0.70 1.40 KREATİNİN mg/dL Kadın 0.6 1.20 T.KOLESTEROL mg/dL <200 HDL-KOLESTEROL mg/dL >35 LDL-KOLESTEROL mg/dL <160 TRİGLİSERİD mg/dL <150 , 43 Hastalarda insülin öncesi ve insülin tedavisi sonrasında beta hücre rezervi değerlendirildi 0 ve 6.hafta sonunda elde edilen veriler toplanarak karşılaştırılarak ve aynı zamanda aralarındaki ilişki değerlendirildi. Hastalarda insülin analoglarının Beta hücre rezervi üzerine olan etkinliği değerlendirildi. Bunun için SPSS 11.0 programında mann-whıtney u ve chı-square testleri kullanıldı. p<0.05 değeri anlamlı olarak kabul edildi. BULGULAR Tablo 7. Her iki grup hastanın cinsiyetlerinin karşılaştırılması CİNSİYET Total KADIN ERKEK N 5 5 10 OAD % 50.0% 50.0% 50.0% N 5 5 10 İNSÜLİN % 50.0% 50.0% 50.0% Tablo 8. Her iki grup hastaların yaşlarının karşılaştırılması G R U P O A D İ N S Ü L İ N YAŞ Ortalama S. Sapma Minimum Maximum N Ortalama S. Sapma Minimum Maximum 46.5000 11.5398 32.00 66.00 10 56.6000 12.4025 38.00 74.00 N 10 P değeri 0.096 Her iki grup hastanın cinsiyet ve yaş dağılımında istatistiksel olarak anlamlı fark bulunmamıştır. Tablo 9. Grup I ve Grup II hastaların proinsülin değerlerinin karşılaştırılması Tedavi Başlangıcı Tedavi sonu 44 PROİNS PROİNS PROİNS PROİNS BAZAL UYARILMIŞ BAZAL UYARILMIŞ GR UP Ortalama 14.9100 24.3500 8.3400 10.9100 S. Sapma 6.1535 9.2969 3.3090 4.1423 Minimum 3.20 8.90 1.80 5.20 Maximum 25.90 37.10 12.50 18.10 N 10 10 10 10 Ortalama 10.6500 15.6300 6.4300 9.9050 S. Sapma 5.1571 6.9811 7.3376 9.3497 Minimum 2.30 3.30 .10 .44 Maximum 17.70 24.30 22.90 30.10 O A D İ N S Ü L İ N N P değeri 10 10 10 10 0.071 0.049 0.174 0.273 Tablo 10. Grup I ve Grup II hastaların c-peptid değerlerinin karşılaştırılması Tedavi Başlangıcı G R U P CPEP BAZAL CPEP UYARILMIŞ Tedavi sonu CPEP BAZAL CPEP UYARILMIŞ 45 Ortalama 0.057 S. Sapma 0.049 Minimum .02 Maximum .19 N 10 Ortalama .1610 S. Sapma .2989 Minimum .01 Maximum .98 O A D İ N S Ü L İ N N P değeri .1670 .1216 .05 .40 10 .3490 .6225 .01 2.06 .1150 .2340 0.09217 .2182 .02 .01 .34 .73 10 10 .7480 1.3340 .7702 1.4137 .19 .39 2.60 4.91 10 10 10 10 0.760 0.909 0.001 0.001 Her iki grup karşılaştırıldığında proinsülin düzeylerinde glukagon verilmesinden 6 dk sonra insülin grubunda anlamlı olarak artmış, diğer ölçümlerde istatistiksel olarak anlamlı fark bulunmamıştır. C-peptid düzeylerinde ise, 6. haftaki ölçümlerin her ikisinde de insülin lehine anlamlı artış görülürken, diğer ölçümlerde anlamlı fark görülmemiştir. Tablo 11. Grup I ve Grup II hastaların HbA1c, BMI, AKŞ, TKŞ değerlerinin karşılaştırılması G R U P O A D İ N S Ü L İ N HbA1C. 0 hafta HbA1c. 6. hafta BMI. 0. hafta BMI. 6. hafta AKŞ 0. hafta TKŞ 0. hafta AKŞ.6. hafta TKŞ.6. hafta Ortalama S. Sapma Minimum Maximum N Ortalama S. Sapma Minimum Maximum 8.1480 1.7701 5.95 10.95 10 10.2790 2.7466 7.25 15.40 6.7930 1.0409 5.23 8.32 10 7.7120 .8021 6.47 8.83 29.1000 5.0211 24.00 42.00 10 29.0500 3.5936 24.00 33.50 28.6000 5.1251 24.00 42.00 10 28.3000 3.5606 24.00 33.00 183.8000 69.7006 105.00 296.00 10 224.2000 60.3817 150.00 327.00 188.8000 81.3877 99.00 291.00 10 215.1000 59.5454 130.00 321.00 127.0000 34.5672 90.00 184.00 10 132.1000 33.4081 90.00 205.00 132.9000 27.6624 96.00 172.00 10 147.6000 39.0447 96.00 226.00 N 10 10 10 10 10 10 10 10 0.082 0.071 0.620 0.790 0.162 0.427 0.650 0.473 P değeri 46 Gruplar karşılaştırıldığında başlangıçta ve 6. haftadaki ölçümlerde hiçbir parametre açısından anlamlı fark bulunmamıştır. Tablo 12.a, b Grup I ve Grup II hastaların Üre, Kreatinin, kolesterol, LDL; HDL; Trigliserit değerlerinin karşılaştırılması Tablo 12 a. G R U P O A D İ N S Ü L İ N URE 0. hafta URE 6. hafta Ortalama S. Sapma Minimum Maximum N Ortalama S. Sapma Minimum Maximum 27.2000 5.1164 17.00 33.00 10 26.3000 7.8038 17.00 38.00 27.4000 6.9314 15.00 40.00 10 27.2000 7.2080 17.00 36.00 .9800 .1619 .90 1.40 10 .9400 .1350 .70 1.10 .8500 .2759 .40 1.20 10 .9200 .1317 .70 1.10 216.0000 34.4867 163.00 281.00 10 211.0000 27.9563 174.00 254.00 N 10 10 10 10 0.570 0.940 0.803 0.591 P değeri KREAT 0. KREAT 6. KOLEST 0. KOLEST 6. hafta hafta hafta hafta LDL 0. hafta LDL 6. hafta 193.9000 20.8244 166.00 227.00 10 199.5000 35.4941 151.00 262.00 128.9000 45.8995 12.00 183.00 10 132.6000 25.0874 98.00 174.00 125.4000 18.9103 93.00 160.00 10 133.6000 37.7542 83.00 198.00 10 10 10 10 0.677 0.940 0.821 0.850 Her iki grubun üre, ktreatinin, kolesterol ve LDL düzeylerinde anlamlı fark bulunmamıştır. Tablo 12 b G R U P O A D İ N S Ü L İ N HDL 0. hafta HDL 6. hafta TG 0. hafta TG 6. hafta Ortalama S. Sapma Minimum Maximum N Ortalama S. Sapma Minimum Maximum 41.7000 10.9245 34.00 73.00 10 36.6000 5.1251 30.00 45.00 41.0000 8.5635 32.00 55.00 10 37.4000 6.5354 29.00 54.00 169.5000 80.5623 57.00 296.00 10 195.6000 85.1237 85.00 395.00 134.0000 58.0383 51.00 232.00 10 148.0000 59.4549 65.00 266.00 N 10 10 10 10 0068 0.395 0.597 0.705 P değeri Her iki grubun HDL ve trigliserit ölçümleri karşılaştırıldığında istatistiksel olarak anlamlı fark görülmemiştir. 47 40 30 20 PROINSILUN 0 Mean +- 2 SE 10 PROINSILUN 1 0 PROINSILUN 2 -10 PROINSILUN 3 N = 10 10 10 10 OAD 10 10 10 10 INSILUN Şekil 4. Gruplar arası proinsülin değerleri 2.5 2.0 1.5 Mean +- 2 SE 1.0 C-PEPTID 0 .5 C-PEPTID 1 0.0 C-PEPTID 2 -.5 N = C-PEPTID 3 10 10 10 10 10 OAD İNSÜLİN 10 10 10 INSULIN OAD Şekil 5. Gruplar arası C-peptid değerleri 48 13 12 11 10 9 Mean +- 2 SE 8 7 Hb A1 C 0 Hb A1 C 6 6 5 N = 10 10 10 OAD 10 INSULIN Şekil 6. Gruplar arası HbA1C değerleri Tablo 13. OAD grubunun pronsülin değerlerinin kendi içinde karşılaştırılması OAD GRUBU PROİNS. TEDAVİ PROİNS. TEDAVİ ÖNC. 0. DK. ÖNC.6. DK. PROİNS. TEDAVİ ÖNC. PROİNS. TEDAVİ SON 0. DK. 0. DK. PROİNS. TEDAVİ PROİNS. TEDAVİ SON ÖNC.6. DK. 6. DK PROİNS. TEDAVİ SON PROİNS. TEDAVİ SON 0. DK. 6. DK ORTALAMA FARK STD. SAPMA P -4.9800 3.2908 0.440 4.2200 3.2908 0.580 5.7250 3.2908 0.319 -3.4750 3.2908 0.718 OAD grubunun kendi içinde karşılaştırılmasında proinsülin değerlerinde anlamlı fark bulunmamıştır. Tablo 14. OAD grubunun C-peptid değerlerinin kendi içinde karşılaştırılması OAD GRUBU C PEPTİD TEDAVİ C PEPTİD TEDAVİ ÖNC. 0. DK. ÖNC.6. DK C PEPTİD TEDAVİ C PEPTİD TEDAVİ ÖNC. 0. DK. SON 0. DK. C PEPTİD TEDAVİ C PEPTİD TEDAVİ ÖNC.6. DK SON 6. DK C PEPTİD TEDAVİ C PEPTİD TEDAVİ SON 0. DK SON 6. DK ORTALAMA FARK STD. SAPMA P -.1880 .3917 0.963 -.5870 .3917 0.449 -.9850 .3917 0.075 -.5860 .3917 0.450 49 OAD grubu kendi içinde karşılaştırıldığında başlangıç ile 6. haftadaki glukagonla uyarılmış değerler arasında anlamlı fark gözlenmemiştir. Tablo 15. OAD grubunun HbA1c değerlerinin kendi içinde karşılaştırılması OAD GRUBUNDA HbA1c TEDAVİ ÖNC. Ortalama S. sapma Minimum Maximum N HbA1c TEDAVİ SON 8.1480 1.7701 5.95 10.95 10 HB A1 C.0 – HB A1 C.6 karşılaştırması: p=0.013 6.7930 1.0409 5.23 8.32 10 OAD grubu kendi içinde karşılaştırıldığında HbA1c düzeyi başlangıca göre istatistiksel olarak anlamlı azalma göztermiştir. Tablo 16. İnsülin grubunun pronsülin değerlerinin kendi içinde karşılaştırılması İNSÜLİN GRUBU PROİNS. TEDAVİ PROİNS. TEDAVİ ÖNC. 0. DK. ÖNC.6. DK. PROİNS. TEDAVİ ÖNC. PROİNS. TEDAVİ SON 0. DK. 0. DK. PROİNS. TEDAVİ PROİNS. TEDAVİ SON ÖNC.6. DK. 6. DK PROİNS. TEDAVİ SON PROİNS. TEDAVİ SON 0. DK. 6. DK ORTALAMA FARK STD. SAPMA P -8.4400 2.7605 0.021 7.5700 2.7605 0.045 13.4400 2.7605 0.0001 -2.5700 2.7605 0.788 İnsülin grubu kendi içinde tedavi öncesi ve sonrasında, 0. ve 6. dakika proinsülin düzeyleri açısından karşılaştırıldığında; tedavi öncesi 0. ve 6. dakikalar, tedavi öncesi 0 ve tedavi sonrası 0. dakikalar ve tedavi öncesi 6. ve tedavi sonrası 6. dakikalar arasında istatistiksel olarak anlamlı fark bulunmuştur. Tablo 17. İnsülin grubunun C-peptid değerlerinin kendi içinde karşılaştırılması İNSÜLİN GRUBU ORTALAMA FARK 50 STD. SAPMA P C PEPTİD TEDAVİ ÖNC. 0. DK. C PEPTİD TEDAVİ ÖNC. 0. DK. C PEPTİD TEDAVİ ÖNC.6. DK C PEPTİD TEDAVİ SON 0. DK C PEPTİD TEDAVİ ÖNC.6. DK C PEPTİD TEDAVİ SON 0. DK. C PEPTİD TEDAVİ SON 6. DK C PEPTİD TEDAVİ SON 6. DK -.1100 0,0605 -0,058 0,0605 0.036 -0,067 0,0605 0.048 -0.1190 0,0605 0.029 0.042 İnsülin grubu C-peptid düzeyleri açısından kendiiçinde karşılaştırıldığında tüm karşılaştırmalar açısından istatistiksel olarak fark saptanmıştır. Tablo 18. İnsülin grubunun HbA1c değerlerinin kendi içinde karşılaştırılması İNSÜLİN GRUBUNDA HbA1c TEDAVİ ÖNC. Ortalama S. sapma Minimum Maximum N HbA1c TEDAVİ SON 10.2790 2.7466 7.25 15.40 10 HB A1 C.0 – HB A1 C.6 karşılaştırması: p=0.005 7.7120 .8021 6.47 8.83 10 İnsülin grubu kendi içinde karşılaştırıldığında HbA1c düzeyi başlangıca göre istatistiksel olarak anlamlı azalma göztermiştir. TARTIŞMA Bizim bu klinik çalışmada bulduğumuz en önemli sonuç, tip 2 diyabetik hastalarda başlangıç tedavisi olarak insülin seçildiğinde sülfonilüre tedavisine kıyasla beta hücre rezervi daha iyi korunmuştur. Bunun yanı sıra her 2 tedavi seçeneği de hastaların metabolik kontrolünü başlangıç düzeylerine göre tedavi sonunda başarılı şekilde sağlamışlardır. Bizim çalışmamızda insülin alan grupta, OAD alan gruba göre, tedavi öncesinde beta hücre reservini gösteren parametreler açısından herhangi bir fark yokken, tedavi sonrasında proinsülin düzeylerinde anlamlı fark tespit edilememiş ancak C-peptid düzeylerinde insülin grubunda OAD grubuna göre istatistiki olarak anlamlı artış gözlenmiştir. Bu artış tedavi sonrasınnda hem bazal C-peptid düzeyleri 51 arasında hem de glukagonla uyarılmış C-peptid düzeyleri arasında saptandı. Tedavi öncesi bazal C-peptid ortalaması OAD grubunda 0.057 ng/mL, insülin grubunda 0.161 ng/mL tespit edilmiş(p>0.05), tedavi sonrasında ise OAD grubunda 0.115 ng/mL, insülin grubunda 0.748 ng/mL bulunmuştur(p=0.001). Metabolik kontrol açısından değerlendirildiğinde ise insülin grubunda HbA1c tedavi başlangıcında % 10.279 iken tedavi sonrasında %7.712’ye düştü ve bu düşüş istatistiksel olarak anlamlı idi(p=0.005). OAD grubunda ise tedavi başlangıcında HbA1c düzeyi tedavi başlangıcında %8.148 iken tedavi sonrasında %6.793’e düştü ve bu düşüş istatistiksel olarak anlamlı idi(p=0.013). Tip 2 diyabetli hastalarda insülin tedavisi ile ilgili olarak uygulanan genel yaklaşım, önce bir oral antidiyabetik kullanıldıktan sonra bu ilaçlara karşı sekonder yetersizlik geliştikten sonra insülin tedavisine başlamak şeklindedir. UKPDS çalışması da diğer tedavi alternatifleri ile insülin tedavisini karşılaştırdığında, insülin lehine bir sonuca ulaşamamıştır(27). Bu gözlemle tip 2 diyabetik hastalarda erken insülin tedavisine başlamaya karşı kullanılan temel argümanları destekler nitelikte olmuştur. Ancak Alvarsson ve arkadaşları(28) yeni tanı konulmuş tip 2 diyabetik hastalarda insülinle oral antidiyabetik tedaviyi karşılaştırdıkları çalışmalarında yukarıda bahsedilen argümanların aksine erken başlanan insülin tedavisinin geleneksel sülfonilüre tedavisine kıyasla gerek endojen insülin sekresyonu, gerekse metabolik kontrol açısından daha üstün olduğunu göstermiştir. Bu çalışmada günde iki kez uygulanan premixinsülin (%30 solubl ve %70 NPH insülin, mixtard 30/70; Novo Nordisk, Cophenhagen, Denmark) kullanlımış olup, bizim çalışmamız bu alanda insülin preperatı olarak 3x lyspro insülin ve 1x NPH insülin protokolü ile özgün bir çalışmadır. Tip 2 diyabetik hastalara insülin tedavisinin gerekliliği ve eğer başlanacaksa zamanı konusundaki tartışmalar yıllardan beri sürmektedir. Tip 1 diyabetik hastalarda mikrovasküler komplikasyonların gelişmesi ve ilerlemesinin altında yatan en önemli faktör yıllar boyu devam eden yüksek glikoz düzeyleridir. Bunun tip 2 diyabet için de doğru olduğu hem epidemiyolojik analizler hem de klinik çalışmalar ile gösterilmiştir(29, 30). Yüksek kan glikoz düzeylerinin makrovasküler hastalık riskini de 52 arttırabileceği gösterilmiştir(31). Geleneksel oral tedavi yöntemleri, glisemik kontrolü arzu edilen düzeylere indirmekte yetersiz kalmakta ve bu amaçla insülin tedavisi kullanılması gerekmektedir. Bunun yanısıra insülin tedavisi ile glisemik kontrolün yanında beta hücre rezervinin korunabilmesi de tip 2 diyabetik hastalarda insülin tedavisinin önemini arttıran bir faktördür. Birkeland ve arkadaşları(32) tip 2 diyabetik hastalarda 42 aylık bir tedavi periyodunda insülin ile OAD’yi karşılaştırdıkları randomize kontrollü çalışmalarında insülin tedavisi ile hipergliseminin kontrolünün daha etkili bir şekilde sağlandığını ve bir kez glisemik kontrol sağlandıktan sonra 42 ay boyunca bir daha bozulmadığını saptamışlardır. Ayrıca bu çalışmada OAD kullanılan hastaların 2/3’ü daha sonradan yetersiz glisemik kontrol nedeniyle insülin tedavisi almak zorunda kalmıştır. Bizim çalışmamızda her iki grup için de takip süresi 6 hafta olup her iki tedavi yöntemiyle de metabolik kontrol sağlanmıştır. Daha sonradan metabolik kontrolde gelişebilecek bozulmaların tayini için bu takip süresi yeterli değildir. Kan glikozu iki yolla elde edilir. Birincisi bağırsaklardan emilen karbonhidratlardan ikincisi ise karaciğerde doğrudan üretimdir. Gerek mevcut, gerekse artmış kan glikoz düzeyleri insülin salınımını uyarır(33). Postprandiyal glikoz salınımını öğünler arasında karaciğerden üretilenden 20-30 kat daha fazladır. Faz 1 insülin salınımı, 10 dakika sürer, hepatik glikoz üretimini baskılar ve faz 2 insülin salınımını kolaylaştırır. Faz 2 insülin salınımı ise 2 saat sürer ve öğün zamanındaki karbonhidratlar üzerine etkilidir. Öğünler arasında çok düşük düzeyde salınan insülin, bazal insülin olarak da adlandırılır ve diğer metabolik ihtiyaçları karşılar. Normal beta hücresi kan glikoz düzeylerine lineer bir biçimde yanıt verir. Bu yanıtın eğrisi açlıktan sonra daha diktir ve yüksek glikoz düzeyleri uzun süre devam ederse bu yanıt eğrisi düzleşir. Bu şekilde glikoz düzeylerine yanıtın kaybına beta hücre yorgunluğu veya glikoz toksisitesi denir(34). Bu durum diyabetin erken dönemlerinde geriye döndürülebilir. Tip 2 diyabette faz 1 salınım yoktur ve faz 2 salınım da gecikmiş veya yetersizdir. Normal insanlarda öğün zamanında görülen insülin salınımındaki keskin pikler tip 2 diyabette gecikmiş, uzamış 53 ve yetersizdir. Tanı ve tedaviden önce beta hücresi insülin direncini kompanse edebilmek için aşırı insülin üretir, fakat sonradan beta hücresinde amiloid biriktiği için insülin üretimi azalır(35). Klinik olarak tip 2 diyabet teşhis edildiğinde normal beta hücre fonksiyonunun %50’si kayba uğramıştır. UKPDS çalışması göstermiştir ki, geriye kalan %50’de diğer tedavi yöntemleriyle yıllar içerisinde azalmaktadır(36). Tüm bu nedenlerle beta hücre fonksiyonunun korunabilmesi tip 2 diyabetin klinik gelişimi açısından hayati öneme sahiptir. Beta hücre fonksiyonu endojen insülinin pulsatil salınım göstermesi ve kısa yarı ömre sahip olması (6-7 dk.) nedeniyle, endojen insülin düzeylerine bakılarak sağlıklı bir şekilde değerlendirilemez. Beta hücre fonksiyonunu değerlendirmede daha uygun bir yöntem endojen insülin üretiminin bir başka göstergesi olan ve daha uzun yarı ömre sahip (30 dk.) C-peptid düzeylerine bakılmasıdır. Bununla birlikte düşük C-peptid düzeyi bulunması, hastada beta hücre fonksiyonu kaybı mı, yoksa geri dönüşümlü glikoz toksisitesi mi olduğunu ayırt etmede yeterince yardımcı olmaz. Diyabetik hastalarda, herhangi bir tedavinin insülin sekresyonu üzerine olan etkisi konusunda hükme varmak için, uygulanan testlerin güvenilir olması ve standardazisyonunun uygun olması gerekmektedir. C-peptid-Glukagon testi insülin sekresyonunu belirlemek amacıyla kullanılan testler arasında en standardize testtir(37). Uzun yıllardır bir çok açıdan geçerliliği kanıtlanmıştır(38-41). Biz de güvenilir olması ve standardizasyonunun tam olarak sağlanmış olması nedeniyle beta hücre rezervinin değerlendirilmesinde glukagonla uyarılmış C-peptid yöntemini çalışmamızda kullandık. C-peptidGlukagon testi, sülfonilüreler veya diğer antidiyabetik ilaçlarla sürdürülen tedaviler esnasında gerçekleştirilir. Bu ilaçlar, aynı zamanda endojen insülin sekresyonunu da etkilerler. Sülfonilüreler ile tedavi edilen hastaların her yıl %5-10’unda sulfonilüre yetersizliği gelişmektedir. Palmer ve arkadaşları(42) beta hücre rezervinin korunmasının diyabetik hastalarda yaşam kalitesi, yaşam beklentisi ve komplikasyonların maliyeti üzerine olası etkilerini inceledikleri çalışmalarında CORE (Center for Outcomes Research) diyabet modeli adını verdikleri, model aracılığı ile sonuçları 54 yorumlamışlardır. Bu modelde bir grup yeni tanı konulan tip 2 diyabetik hasta tıpkı UKPDS çalışmasındaki gibi HbA1C de tipik bir artış göstermiş, modellenen diğer grupta ise beta hücre fonksiyonu stabil kalacak şekilde HbA1c sabit olarak kurgulanmıştır. Buna göre pankreatik beta hücre fonksiyonu sabit tutulduğunda 50 yıllık bir zaman diliminde tip 2 diyabetik hastaların yaşam beklentisi ve yaşam kalitesi artmakta, buna karşılık komplikasyonların maliyetleri azalmaktadır. Tip 2 diyabet ilerleyici bir hastalıktır. Tanı konulduğunda yıllar öncesinden hastalığın başladığı bilinmektedir. ABD’de halihazırda tanı konulmuş olan 10 milyon hastanın yanında 5 milyon da tanı konulmamış tip 2 diyabetiğin var olduğu tahmin edilmektedir(43). Şurası bilinmektedir ki, tip 2 diyabet saptandığında hastaların çoğu son 5-10 yıldır hiperglisemik seyretmekte ve mikro-makrovasküler komplikasyonlar gelişmiştir(44). Örneğin; Harris ve arkadaşları(45) yaptıkları klinik çalışmada göstermişlerdir ki, diyabet tanısı konulmadan yıllar önce dahi retinopati gelişmektedir. ABD ve Avusturalya toplumlarında tanı anında retinopati sıklığını araştıran Harris ve arkadaşları bu sıklığı, ADB’de %20.8, Avusturalya’da %9.9 bulmuşlardır. Retinopati prevelansı ile diyabet süresi arasında lineer bir ilişki bulunduğundan dolayı bu analizden yola çıkarak araştırıcılar Amerikan diyabetiklerinde tanıdan 6.5 yıl önce, Avusturalya’lı diyabetiklerde ise tanıdan 4.2 yıl önce diyabetin başladığını tahmin etmişlerdir. Tanı anında diyabetik hastaların yaklaşık %50’sinde diyabete bağlı organ hasarı gelişmekte(46), dahası aşikar hiperglisemi gelişene kadar beta hücrelerinin sekretuar fonksiyonları şiddetli bir şekilde hasara uğramıştır(47). Diyabetin etiyolojisi, patogenezi ve ilerleyici yapısını göz önünde bulunduran araştırıcılar, yeni tanı konulmuş tip 2 diyabetik hastalara erken girişimde bulunulmasının beta hücre fonksiyonundaki ilerleyici kaybı önleyip önlemediğini ve elde edilen terapötik yanıtın sürdürülebilirliğinin mümkün olup olmadığını belirlemek amacıyla klinik çalışmalar yürütmektedirler. 55 Tip 2 diyabeti tedavi etmek amacıyla her biri farklı etki mekanizmasına sahip birçok oral ajan mevcuttur. Ancak uzun dönemli çalışmalar oral ajanların kan glikoz düzeylerini kontrol etmekte başarısız olduklarını göstermiştir. UKPDS den alınan 6 yıllık bir çalışmada yeni tanı konulmuş tip 2 diyabetik hastalar sülfonilüre, metformin veya insülin tedavileri almak üzere randomize edilmiştir(48). Yine UKPDS’den alınan bir başka çalışmada ise 9 yıllık bir süreç boyunca tek başına diyet, insülin, sülfonilüre ve metformin tedavileri karşılaştırılmıştır(49). Her bir terapötik ajan sadece diyet alan grupla karşılaştırıldığında hepsi de HbA1c’yi %7’nin altına düşürme açısından 2-3 kat başarılı olmuşlarsa da bu kontrol zamanla bozulmuştur. Çalışmanın 3. yılında hastaların %50’si monoterapi ile bu hedefe ulaşmışlarsa da 9. yılda bu oran %25’e düşmüştür. Sonuç olarak oral ajanların geleneksel kullanımı özellikle de sülfonilüreler orta derecede insülin sekresyonuna sahip hastalarda ilk basamakta kullanım açısından bir takım soru işaretleri taşımaktadır. Sülfonilüreler hızlı bir glisemik kontrol sağlamalarına rağmen yıllar sonra etkinlikleri azalmaktadır(50). Ayrıca bu ajanlar pankreas üzerinde uzamış insülinotropik etkiye sahip oldukları için prematür beta hücre yetersizliğini de indüklemektedirler. UKPDS çalışmasında sülfonilüre ile tedavi edilen hastalarda beta hücre fonksiyonu ilk yılda artmasına rağmen, daha sonradan hızla düşmüştür(50). İlkova H ve arkadaşlarının (64) yaptığı bir çalışmada yeni tespit edilmiş diyete cevap vermeyen 13 tip 2 diyabetli hastaya 2 hafta boyunca devamlı yoğun insülin tedavisi uygulanmış, bu hastalardan 9’unda daha sonra herhangi bir ilaç kullanılmadan yaklaşık 50 ay boyunca glisemik kontrol sağlanmıştır. Bunun sonucunda diyetle tedaviye cevap vermeyen tip 2 diyabetli hastaların önemli bir kısmında kısa dönem yoğun insülin tedavisi sonucu etkin bir cevap oluşmuştur. İlaç tedavisine gerek kalmaksızın uzun dönem etkin glisemik kontrol sağlanmıştır. İnsülin direncini kompanse etmek amacıyla pankreatik beta hücreleri daha fazla insülin üretme yetenekleri hiperglisemi gelişimini etkilemektedir. Son yıllarda yapılan çalışmalar göstermiştir ki; beta hücre fonksiyonu diyabetin erken dönemlerinde hatta bozulmuş glikoz toleransı döneminde bile 56 azalmaktadır (51, 52). Hastalığın gelişmesinden sonraki erken dönemlerde insülin tedavisi başlanması endojen insülin sekresyonunun korunması açısından iyi bir seçenektir. Hızlı etkili insülin analogları, örneğin lispro insülin yemekten 15 dk. önce uygulanır ve hızla etkiyerek postprandiyal kan glikozu düzeylerini kontrol eder. Bu etkileri ile normal insülin salınımını taklit eder. İnsülin lispro, tip 2 diabette öğün zamanı kan şekeri kontrollerini daha iyi sağlamak amacıyla son zamanlarda geliştirilen bir ajandır. İnsülin lispro enjeksiyon bölgesinde hızla monomerik formuna disosiye olur ve böylece, hızlı bir absorbsiyon oranı ve kısa bir etki süresine sahiptir(53). Human reguler insülin ile kıyaslandığında, onun yarısı kadar bir sürede 2 kat daha fazla pik düzeye ulaşır, ayrıca etki de regüler insülinden daha kısadır(53,54). Bu nedenle insülin lispro tedavisi ile daha çok postpirandial kan şekeri yükseklikleri hedeflenmiş olur. Lispro insülinle ilgili olarak günümüze değin birçok klinik çalışma gerçekleştirilmiş olmasına rağmen bunların içinde çokuluslu ve çok merkezli olarak yapılan 4 büyük çalışma mevcuttur(55-58). Bu çalışmalarda toplam 2300 diabetik hasta yeralmış ve insülin lispro ile öğün zamanı glisemik kontrol incelenmiştir. Bu çalışmalar hem paralel hemde çapraz karşılaştırmalı olarak dizayn edilmişlerdir. Çalışmaların süreleri 6-12 ay arasında değişmektedir. İnsülin lispro yemekten önceki 15 dakika içinde ve regüler insülin ise yemekten 30-45 dakika önce uygulandı. Kan şekeri konsantrasyonları, yemekten önce ve yemekten 1-2 saat sonra değerlendirildi. Benzer dozlarda, postpirandial plazma glukoz düzeyleri regüler insüline kıyasla lispro insülin uygulanan hastalarda 27-45 mg/dL daha düşük bulunmuştur. Daha küçük çaplı birtakım çalışmalarda da devamlı subkutan insülin infüzyonu (CSII) tedavisinde insülin lispro kullanılmış ve benzer bulgular bulunmuştur(59-63). Forst ve arkadaşları(64) erken tip 2 diyabetik hastalarda insülin lispro ile oral antidiyabetiği etkinlik ve güvenlik açısından karşılaştırılmışlardır. 143 hastayı dahil ettikleri bu çok merkezli çalışmalarında glukagonla uyarılmış C-peptid düzeyleri en az 0.4 nmol/L olan hastalar 26 hafta boyunca lispro veya OAD almışlardır. Lispro tedavisi ile postprandiyal kan glikozu kontrolü daha iyi sağlanmıştır. 57 Bizim çalışmamızda insülin grubunda proinsülin seviyeleri başlangıca göre düşüş göstermiiştir. Ancak bu düşüş OAD grubunda yoktur. Bu sonuca göre analog insülin tedavisi ile uygulanan intensif insülin tedavisinin yeni başlangıçlı tip 2 diyabetiklerde beta hücre rezervini iyileştirdiği düşünülebilir. Ancak bu durum OAD alanlarda gözlenmemektedir. Bu nedenle yeni tanı konulan hastalarda analog insülinler iyi bir seçenektir. Alvarsson ve arkadaşları(28) bizim çalışmamızla benzerlik gösteren ancak premix insülin ile sülfonilüreyi karşılaştırdıkları çalışmalarında beta hücre fonksiyonu, glisemik kontrol ve yaşam kalıtesini değerlendirmişlerdir. Çalışmaya adacık hücre antikoru negatif olan erken tip 2 diyabetik 39 hasta dahil edilmiş olup, çalışma 2 yıldan uzun sürdürülmüştür. Çalışmada beta hücre rezervi bazal ve glukagonla uyarılmış C-peptid ve proinsülin düzeylerine göre değerlendirilmiştir. Çalışmada varılan iki ana sonuçtan birincisi, beta hücre fonksiyonu parametrelerinin insülin grubunda daha iyi korunduğu, ikincisi ise başlangıçta metabolik kontroldeki iyileşme benzer düzeylerde sağlanmışken, insülin bu kontrol süreklilik arz etmiş oysa OAD grubunda metabolik kontrol gittikçe bozulmuştur. İnsülin tedavisinin hiperlipidemiye yol açtığı şeklinde genel bir kanı vardır(28). Bununla birlikte biz çalışmamızda insülin tedavisinin lipid düzeylerini arttırmadığı sonucuna ulaştık. Bizim çalışmamızda da lipid düzeyleri gerek insülin grubunda gerekse OAD grubunda başlangıca kıyasla tedavi sonunda anlamlı değişiklikler göstermemiştir. Yine kilo alımı açısından da insülin tedavisi, OAD tedavisi alanlardan farklılık göstermemiştir. Bunun olası nedeni erken tanı konan diyabetiklerde hastaların ihtiyaç duydukları ve uygulanan insülin dozlarının hastalığın ileri dönemlerinde kullanılan insülin dozlarından daha az olmasıdır. Bizim çalışmamızda da BMI açısından da tedavi öncesi ve sonrası arasında anlamlı değişiklikler kaydedilmemiştir. Araştırıcılar bu sonuçlarla tip 2 diyabetik hastalarda başlangıç tedavisi olarak insülin seçildiğinde endojen insülin sekresyonunun ve metabolik kontrolün geleneksel sülfonilüre tedavisine kıyasla daha iyi korunabildiği kanaatine ulaşmışlardır. Bizim çalışmamızda elde ettiğimiz sonuçlar Alvarsson ve arkadaşlarının sonuçları ile uyumluluk göstermektedir. 58 Sonuç olarak; tip 2 diyabetik hastalarda başlangıç tedavisi olarak analog insülin seçildiğinde sülfonilüre tedavisine kıyasla beta hücre rezervi daha iyi korunmuştur. Bunun yanı sıra her 2 tedavi seçeneği de hastaların metabolik kontrolünü başlangıç düzeylerine göre tedavi sonunda başarılı şekilde sağlamışlardır. Bu nedenle yeni tanı konulan tip 2 diyabetik hastalarda analog insülinlerle uygulanan intensif insülin tedavisi iyi bir seçenektir. KAYNAKLAR 59 1. Altuntaş Y. Diabetes mellitus’un tanımı, tanısı ve sınıflandırması in: Yenigün M, Altuntaş Y(eds): Her Yönüyle Diabetes Mellitus. Nobel Tıp kitabevi, İstanbul 2001; (1):51-63. 2. Yenigün M. Diabetes mellitus fizyopatolojisi in: Yenigün M, Altuntaş Y(eds): Her Yönüyle Diabetes Mellitus. Nobel Tıp kitabevi, İstanbul 2001; (1):85-129. 3. Altuntaş Y. Tip 2 diabetes mellitus’un patogenezi in: Yenigün M, Altuntaş Y(eds): Her Yönüyle Diabetes Mellitus. Nobel Tıp kitabevi, İstanbul 2001; (1):219-237. 4. Dahl-Jorgensen K, Brinchmann-Hansen O, Hanssen KF et al. Effect of near normoglycaemia for two years on progression of early diabetic retinopathy, nephropathy, and neuropathy: The Oslo Study. BMJ 1986; 293: 1195-1199. 5. The Diabetes Control and Complications Trial (DCCT) Research Group. The effects of intensive treatment of diabetes on the development and progression of long-term complications in insulindependent diabetes mellitus. N Engl J Med 1993; 329: 977-986. 6. Reichard P, Nilsson B-Y, Rosenquist U. The effect of long-term intensified insulin treatrnent on the development of microvascular complications of diabetes mellitus. N Engl J Med 1993; 329: 304-309. 7. Wang PH, Lou 1, Chairners TC. Meta-analysis of effects af intensive blood glucose can trol on late corn plications of type 1 diabetes. Lancet 1993; 341: 1306-1309. 8. Stephenson J, Fuller JH, on behalf of EURODIAB IDDM Complications Study Group. Microvascular and acute complications in IDDM patients: the EURODIAB IDDM Complications Study. Diabetologia 1994; 37: 278-285. 9. Krolewski AS, Laffel LMB, Krowlewski M, Quinn M, Warram JH. Glycosylated haemoglobin and the risk of microalbuminuria in patients with insulin-dependent diabetes rnellitus. N Engl J Med 1995; 332: 1251-1255. 60 10. Uusitupa MIJ, Niskanen LK, Siitonen O, Voutilainen E, Pyörala K. Ten-year cardiovascular mortality in relation to risk factors and abnormalities in lipoprotein composition in Type 2 (non insulin dependent) diabetic and non diabetic subjects. Diabetologia 1993; 36: 1175-1184. 11. Kuusisto J, Mykkanen L, Pyörala K, Laakso M. NIDDM and its metabolic control predict coronary heart disease in elderiy subjects. Diabetes 1994; 43: 960-967. 12. Binder C. Absorption of injected insulin: a clinical-pharmacological study. Acta Pharmacol Toxicol 1969; (Suppl 2) 27:1-84. 13. Gallaway JA, Spradlin CT, Nelson RL et al. Factors influencing the absorption, serum insulin concentration and blood glucose responses after injections of regular insulin and various mixtures. Diabetes Care 1981; 4: 366-376. 14. American Diabetes Association(ADA). Clinical Practice Recommendations. Position statement: insulin administration. Diabetes Care 1995;18 (1 Suppl):29-32. 15. Lean MEJ, Ng LL, Tennison BR. Interval between insulin injection and eating in relation to blood glucose control in adult diahetics. BMJ 1985; 290: 105-108. 16. Jorgensen LN, Nielsen FS. Timing of premeal insulins in diabetic patients on a multiple daily injection regimen. A questionnaire study. Diabetologia 1990; 33: A 116. 17. Ciszak E, Beals JM, Baker JC, Carter ND, Frank BM, Smith GD. Role of C-terminal B-chain residues in insulin assembly: The structure of hexameric Lys(B28), Pro(B29)-human insulin. Structure 1995; 3: 615-622. 18. Brems DN, Alter LA, Beckage MJ, Chance RE, DiMarchi RD, Green LK. Altering the association properties of insulin by amino acid replacement. Protein Eng 1992; 5(6): 527-533. 19. Howey DC, Bowsher RR, Brunelle RL, Woodworth JR. [Lys(B28), Pro(B29)]-human insulin. A rapidly absorbed analogue of human insulin. Diabetes 1994; 43: 396-404. 61 20. Baker EN, Blundell TL, Cutfield JF, Cutfield SM, Dodson EJ, Dodson GG. The structure of 2 Zn pig insulin crystals at 1.5 A resolution. Philos Trans R Soc Lond B Biol Sci 1988; 19: 369-456. 21. Brange J. Insulin preparations. In: Brange J, et al. Galenics of insulin: The “physico-chemical” and pharmaceutical aspects of insulin and insulin preparations. Berlin, Springer-Verlag 1987; 17-31 22. Joseph SE, Korzon-Burakowska A, Woodworth JR, Evans M, Hopkins D, Janes JM, Arniel SA. The action profile of lispro is not blunted by rnixing in the syringe with NPH insulin. Diabetes Care 1998; 21(12): 2098-102. 23. Clore I, Woodworth IR, Cerirnele BJ, Blackard WG, Kurtz D. Mixing insulin lispro (LP) with ultralente (U) insulin does not result in decreosed or delayed LP absorption. Diabetes 1997; 46(1 Suppl): 150A. 24. Feingios MN, Thacker GH, English J, Bethei MA, Lane JD. Modification of postprandial hyperglycemia with Insulin lispro improves glucose control in patients with type 2 diabetes. Diabetes Care 1997; 20(10): 1539-1542. 25. Bastyr EJ, Johnson ME, Trautmann ME, Anderson JH, Vignati L. İnsulin iispro in the treatment of patients with type 2 diabetes meiiitus after oral agent failure. Ciinical Theropeutics in press 1999; 11:35-39. 26. Brodows R, .Schwortz S, .Stewart G, Zagar A, Graf G, Bastyr E. Combination of insulin Lispro (LP), Metformin (MF), or Bedtime NPH (NPH) with su/fonylurea (SU) following secondary SU failure. Diabetes 1999; 48(Supp 1): A 104. 27. UK Prospective Diabetes Study: Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet 352:837–853, 1998 28. Alvarsson M, Sundkvist G, Lager I, Henricsson M, Berntorp K, Fernqvist-Forbes E, Steen L, Westermark G, Westermark P, Orn T, Grill V. Beneficial effects of insulin versus sulphonylurea on 62 insulin secretion and metabolic control in recently diagnosed type 2 diabetic patients. Diabetes Care. 2003 Aug;26(8):2231-7. 29. UK Prospective Diabetes Study (UKPDS) Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet 1998; 352(9131): 854-61. 30. Ohkubo Y, Kishikawa H, Araki E et al. Intensive insulin therapy prevents the progression of diabetic microvascular complications in Japanese patients with non-insulin-dependent diabetes mellitus: a randomized prospective 6-year study. Diabetes Res Clin Pract 1995; 28: 103-17. 31. Laakso M. Glycemic control and the risk for coronary heart disease in patients with non-insulindependent diabetes mellitus. The Finnish studies. Ann Intern Med 1996;124:127-30. 32. K. I. Birkeland , U. Rishaug , K. F. Hanssen A2, S. Vaaler A2 NIDDM: a rapid progressive disease Results from a long-term, randomised, comparative study of insulin or sulphonylurea treatment. Diabetologia (1996) 39: 1629-1633 33. Kahn SE. The importance of the beta-cell in the pathogenesis of type 2 diabetes mellitus. Am J Med 2000;108(suppl 6a):2S-8S. 34. Glaser B, Cerasi E. Early intensive insulin treatment for induction of long-term glycaemic control in type 2 diabetes. Diabetes Obes Metab 1999;1:67-74. 35. Polonsky KS, Given BD, Hirsch LJ, Tillil H, Shapiro ET, Beebe C, et al. Abnormal patterns of insulin secretion in non-insulin-dependent diabetes mellitus. N Engl J Med 1988;318:1231-9. 36. U.K. prospective diabetes study 16. Overview of 6 years’ therapy of type II diabetes: a progressive disease [published correction appears in Diabetes 1996;45:1655]. Diabetes 1995;44:1249-58. 37. Faber OK, Binder C: C-peptide response to glucagon: a test for the residual beta-cell function in diabetes mellitus. Diabetes 26:605–610, 1977 63 38. Madsbad S, Sauerbrey N, Moller-Jensen B, Krarup T, Kuhl C: Outcome of the glucagon test depends upon the prevailing blood glucose concentration in type I (insulin-dependent) diabetic patients. Acta Med Scand 222:71–74, 1987 39. Gottsater A, Landin-Olsson M, Fernlund P, Gullberg B, Lernmark A, Sundkvist G: Pancreatic betacell function evaluated by intravenous glucose and glucagon stimulation: a comparison between insulin and C-peptide to measure insulin secretion. Scand J Clin Lab Invest 52:631–639, 1992 40. Gjessing HJ, Reinhold B, Pedersen O: The effect of chronic hyperglycaemia on the islet B-cell responsiveness in newly diagnosed type 2 diabetes. Diabet Med 9:601–604, 1992 41. Berger B, Stenstrom G, Sundkvist G: Random C-peptide in the classification of diabetes. Scand J Clin Lab Invest 60:687–693, 2000 42. Andrew J. Palmera, Stéphane Rozea, William J. Valentinea, Michael E. Minshallb, Morten Lammerta, Alan Oglesbyc, Clarice Hayesc and Giatgen A. Spinasd What Impact Would Pancreatic Beta-cell Preservation Have on Life Expectancy, Quality-adjusted Life Expectancy and Costs of Complications in Patients with Type 2 Diabetes? A Projection Using the CORE Diabetes Model CURRENT MEDICAL RESEARCH AND OPINION® VOL. 20, SUPPL 1, 2004, S59–S66 43. American Diabetes Association. Diabetes Facts and Figures. http://www.diabetes.org/ada/facts.asp. Accessed November 21, 2000. 44. UK Prospective Diabetes Study Group. Cost effectiveness analysis of improved blood pressure control in hypertensive patients with type 2 diabetes: UKPDS 40. BMJ 1998; 317:720-6. 45. UK Prospective Diabetes Study Group. Cost effectiveness of an intensive blood glucose control policy in patients with type 2 diabetes: economic analysis alongside a randomised controlled trial. UKPDS 41. BMJ 2000; 320:1373-8. 64 46. United Kingdom Prospective Diabetes Study Group. UK Prospective Diabetes Study 6. Complications in newly diagnosed type 2 diabetic patients and their association with different clinical and biochemical risk factors. Diabetes Res. 1990;13:1-11. 47. Polonsky KS, Sturis J, Bell GI. Seminars in medicine of the Beth Israel Hospital, Boston. Noninsulin-dependent diabetes mellitus -- a genetically programmed failure of the beta cell to compensate for insulin resistance. N Engl J Med. 1996;334:777-783. 48. United Kingdom Prospective Diabetes Study Group. United Kingdom Prospective Diabetes Study 24: a 6-year, randomized, controlled trial comparing sulfonylurea, insulin, and metformin therapy in patients with newly diagnosed type 2 diabetes that could not be controlled with diet therapy. Ann Intern Med. 1998;128:165-175. 49. Turner RC, Cull CA, Frighi V, Holman RR. Glycemic control with diet, sulfonylurea, metformin, or insulin in patients with type 2 diabetes mellitus: progressive requirement for multiple therapies (UKPDS 49). UK Prospective Diabetes Study (UKPDS) Group. JAMA. 1999;281:2005-2012. 50. United Kingdom Prospective Diabetes Study Group. UK prospective diabetes study 16. Overview of 6 years' therapy of type II diabetes: a progressive disease. Diabetes. 1995;44:1249-1258. 51. Weyer C, Bogardus C, Mott DM, Pratley RE. The natural history of insulin secretory dysfunction and insulin resistance in the pathogenesis of type 2 diabetes mellitus. J Clin Invest. 1999;104:787-794. 52. Kahn SE. The importance of the beta-cell in the pathogenesis of type 2 diabetes mellitus. Am J Med. 2000;108(suppl 6a):2S-8S. 53. Bowsher RR, Brunelle RL, Woodworth JR. [Lys(B28), Pro(B29)]-human insulin. A rapidly absorbed analogue of human insulin. Diabetes 1994; 43: 396-404. 54. Tuominen JA, Karonen SL, Melamies L, Bolli G, Koivisto VA. Exercise induced hypoglycemia in IDDM patients treated with a short acting insulin analogue. Diabetologia 1995; 38: 106-111. 65 55. Anderson JH jun. Brunele RL, Koivisto VA, Trautmann ME, Vignati L, DiMarchi R. Improved mealtime treatment of diabetes mellitus using an insulin analoge. Multicente insulin Lispro Study Group. Clin ther 1997; 19: 62-72. 56. Anderson JH jun. Brunelle RL, Koivisto VA, et al. Reduction of postprandial hyperglycemia and frequency of hypoglycemia in IDDM patients on insulin analog treatment. Diabetes 1997; 46: 265-70. 57. Anderson JH jun. Brunelle RL, Keohane P, et al. Insulin analoge improves mealtime treatment of NIDDM patients. Arch Intern Med 1997; 157: 1249-55. 58. Holcombe J, Zalani S, Arora V. Comparative study of insulin lispro and regular human insulin in 481 adolescants with type 1 diabetes. Diabetologia 1997; 40: suppl 1, A344. 59. Zinman B, Tildesley H, Chiasson J-L, Tsui E, Strack T. Insulin lispro in CSII: results of a doubleblind cross-over study. Diabetes 1997; 46: 440-43. 60. Hanaire H, Bringer J, Lassman-Vaque V, et al. Improvenet of HbA1c without increasing hypoglycemia risk in diabetic patients treated with insulin lispro in external pumps. Diabetologia 1997; 40: suppl 1, A10. 61. Garg SK, Anderson J, Gerard L, Chase P. Impact of Humalog on HbA1c values in insulin pump users. Diabetes 1999; 48: suppl 1, A224. 62. Melki V, Renard E, Lassmann-Vague V, et al. Improvenet of HbA1c and blood glucose stability in IDDM patients treated with lispro insulin analog in external pumps. Diabetes Care 1998; 21: 977-82. 63. Forst T, Eriksson JW, Strotmann HJ, Bai S, Brunelle R, Gulliya KS, Gack S, Gudat U. Metabolic effects of mealtime insulin lispro in comparison to glibenclamide in early type 2 diabetes. Exp Clin Endocrinol Diabetes. 2003 Apr;111(2):97-103. 64. Ilkova H, Glaser B, Tunckale A, Bagriacik N, Cerasi E. Induction of long-term glycemic control in newly diagnosed type 2 diabetic patients by transient intensive insulin treatment. Diabetes Care. 1997 Sep;20(9):1353-6. 66